Current Issue
2026, Volume 40, Issue 8
 PDF
PDF  Cited by
Cited by HTML
HTML
2026,
40(8): 080102.
doi: 10.11858/gywlxb.20261102
Abstract:
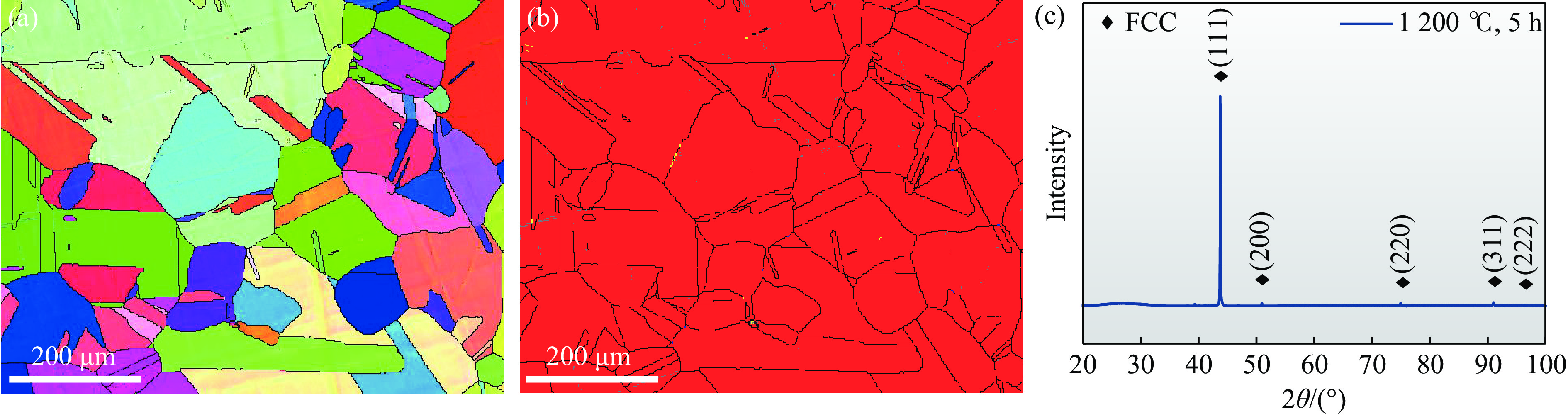

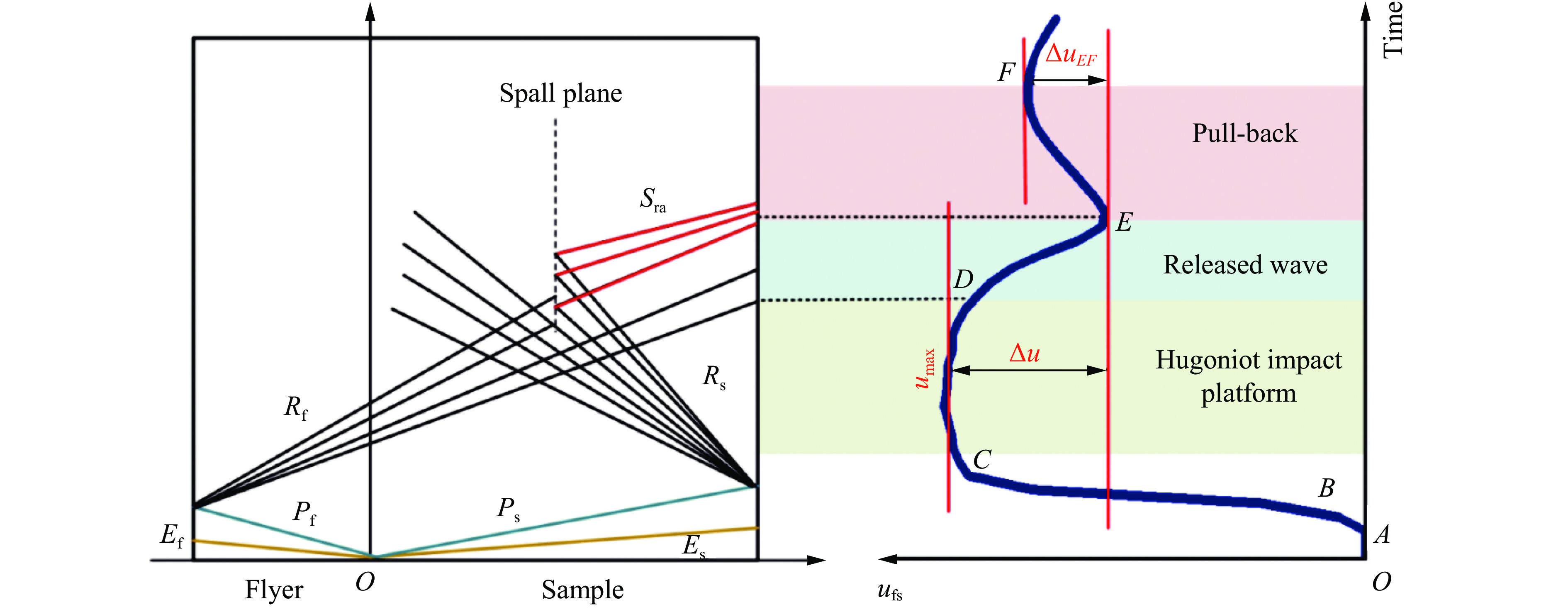
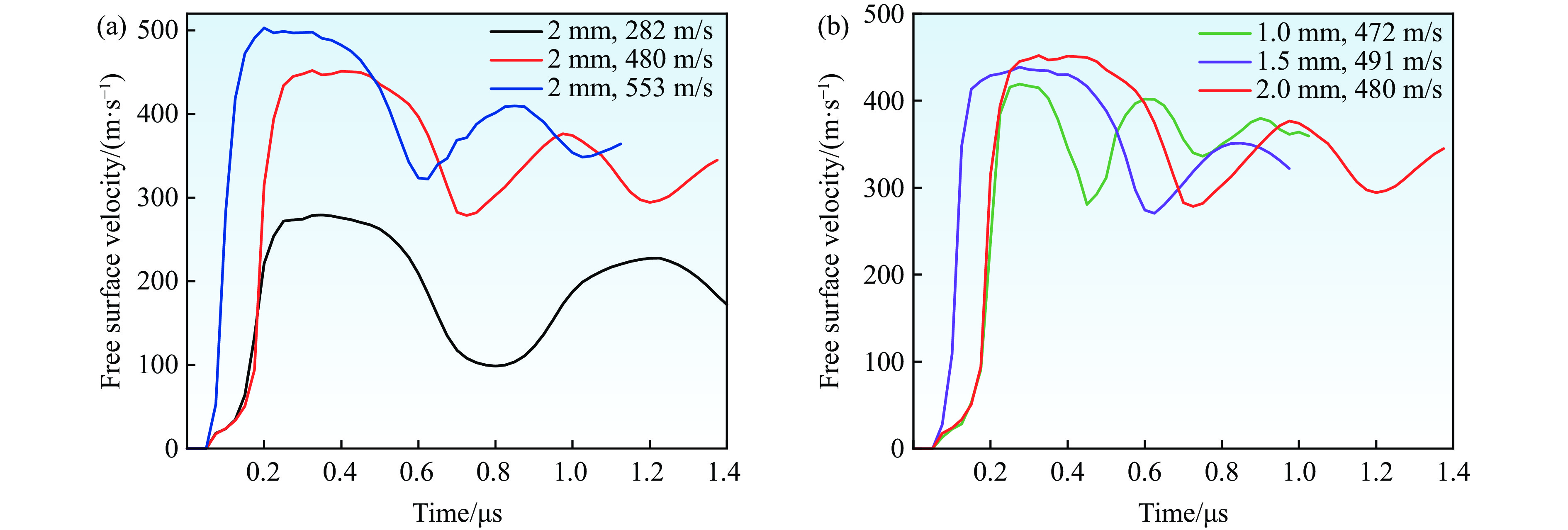


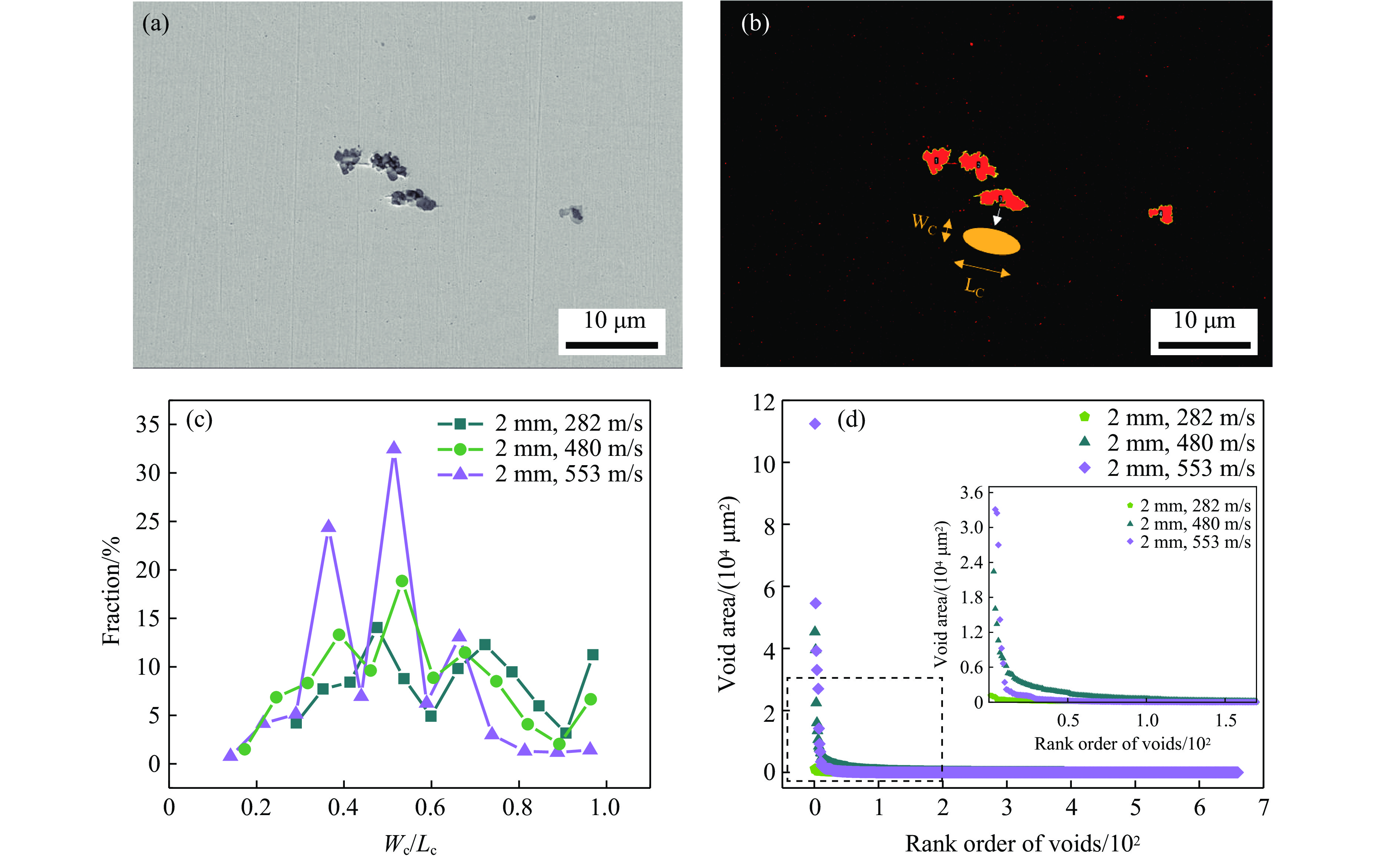


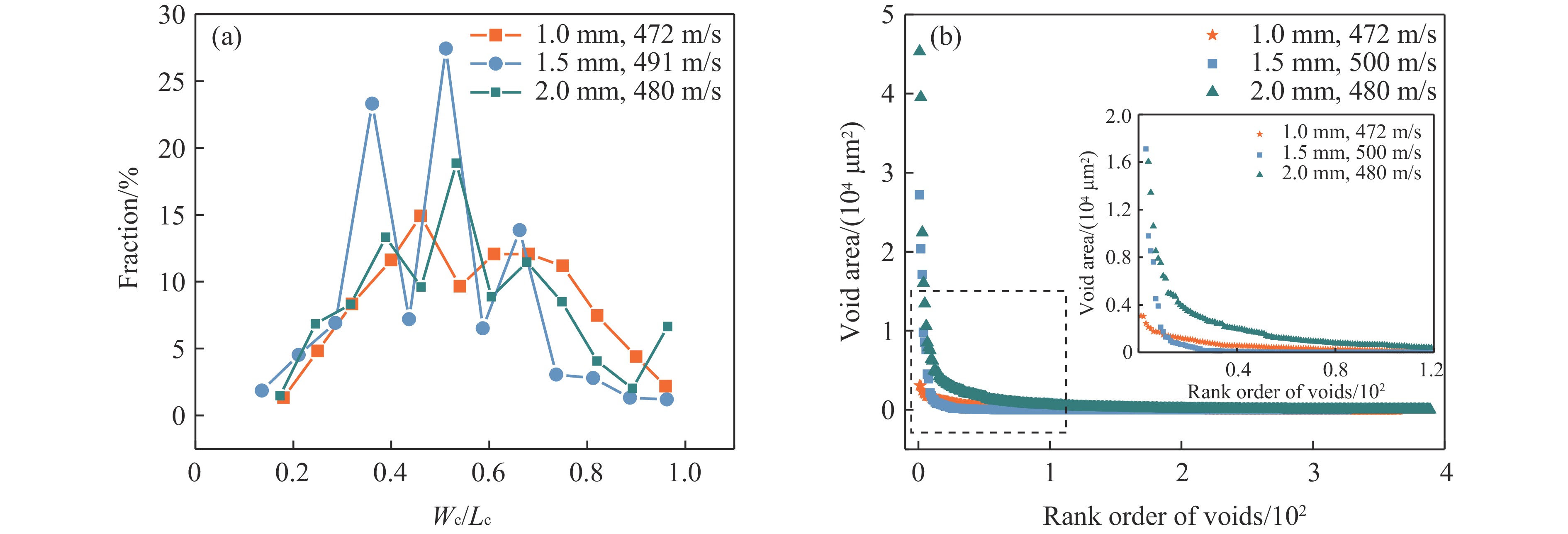

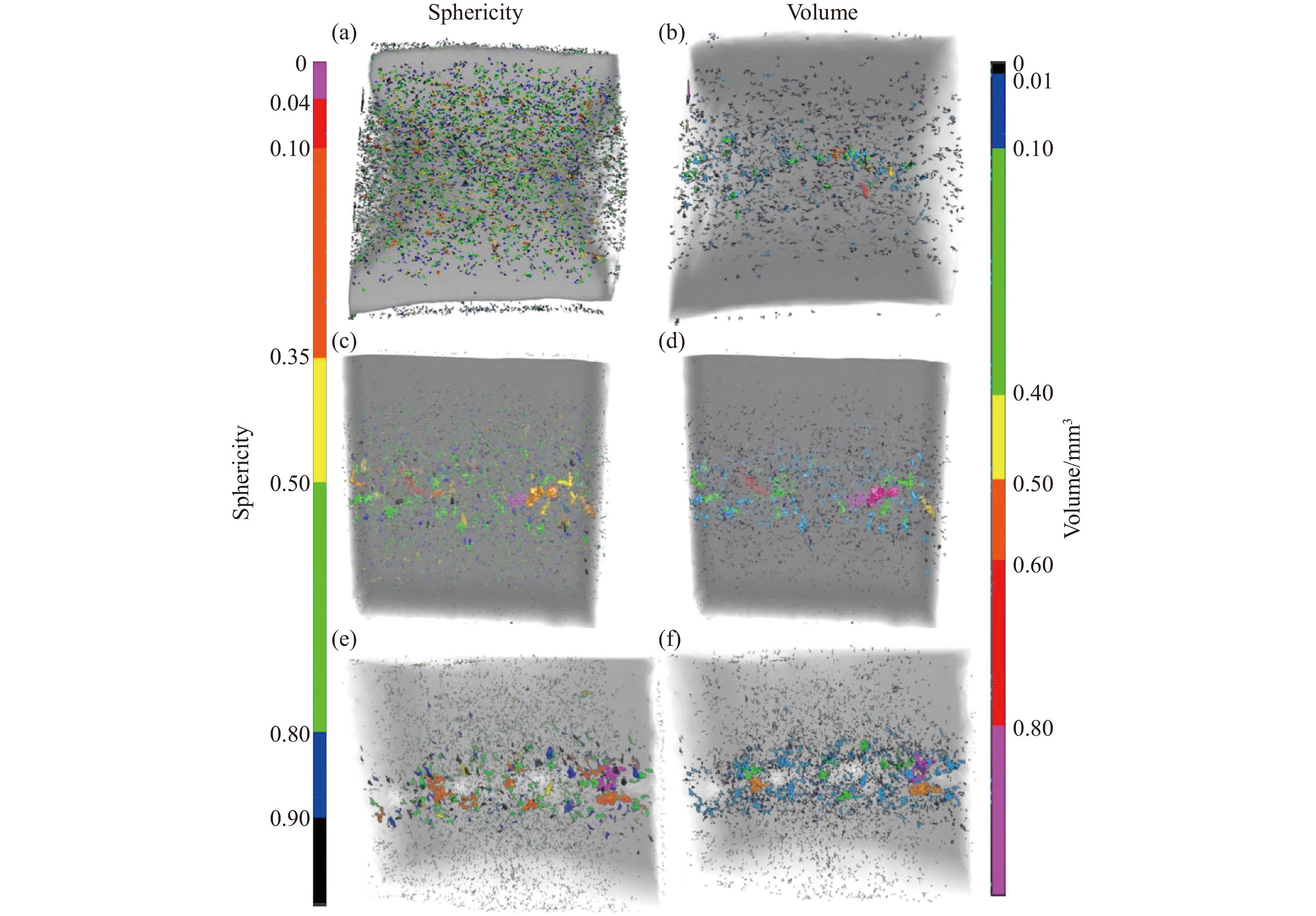
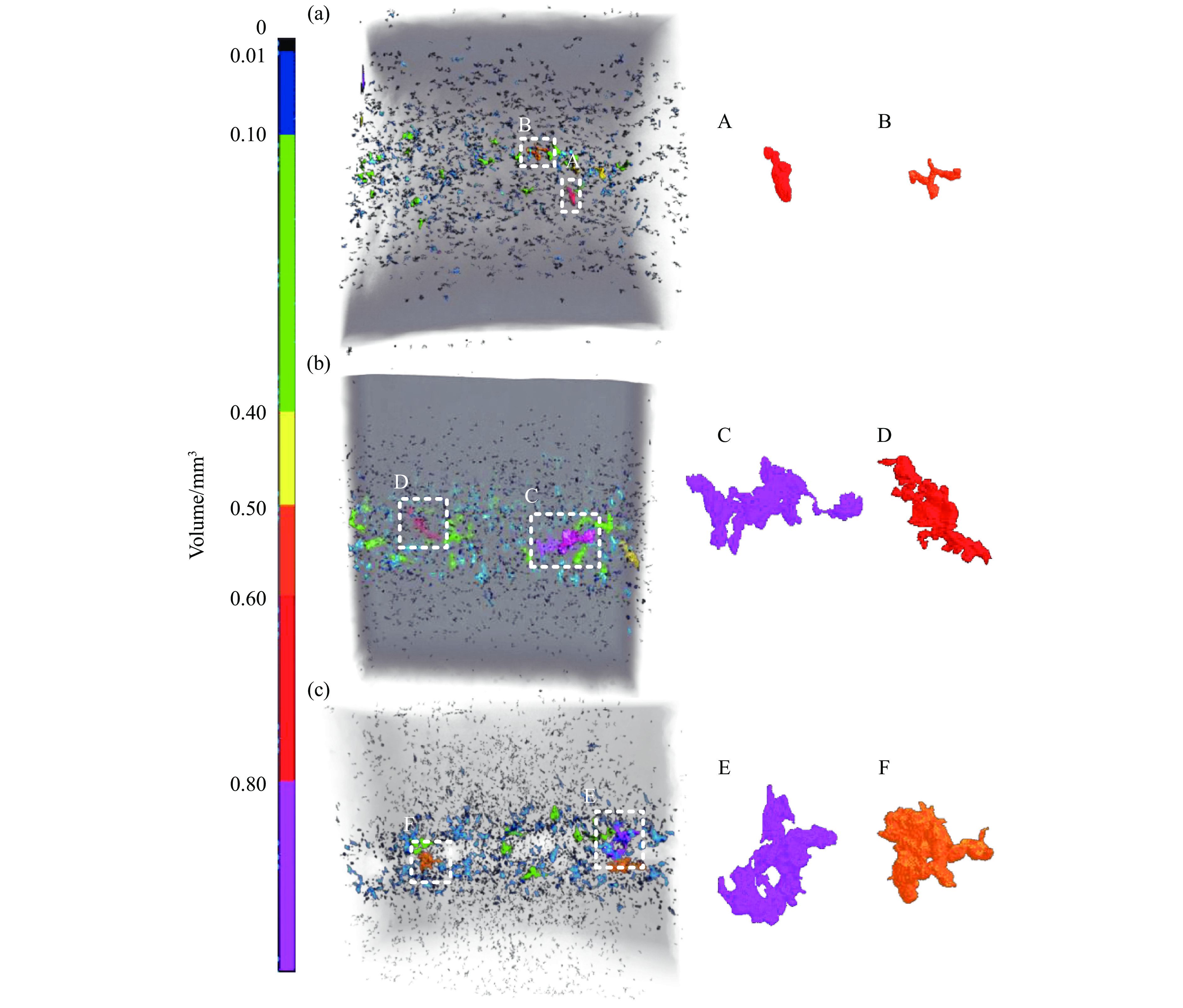
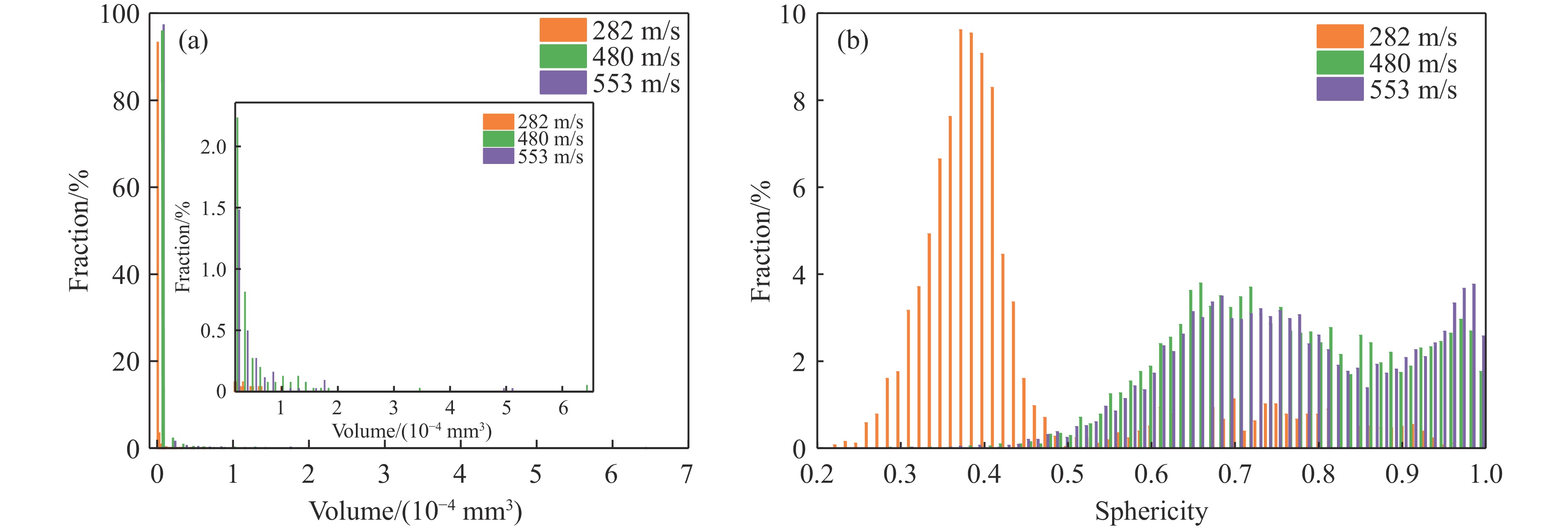
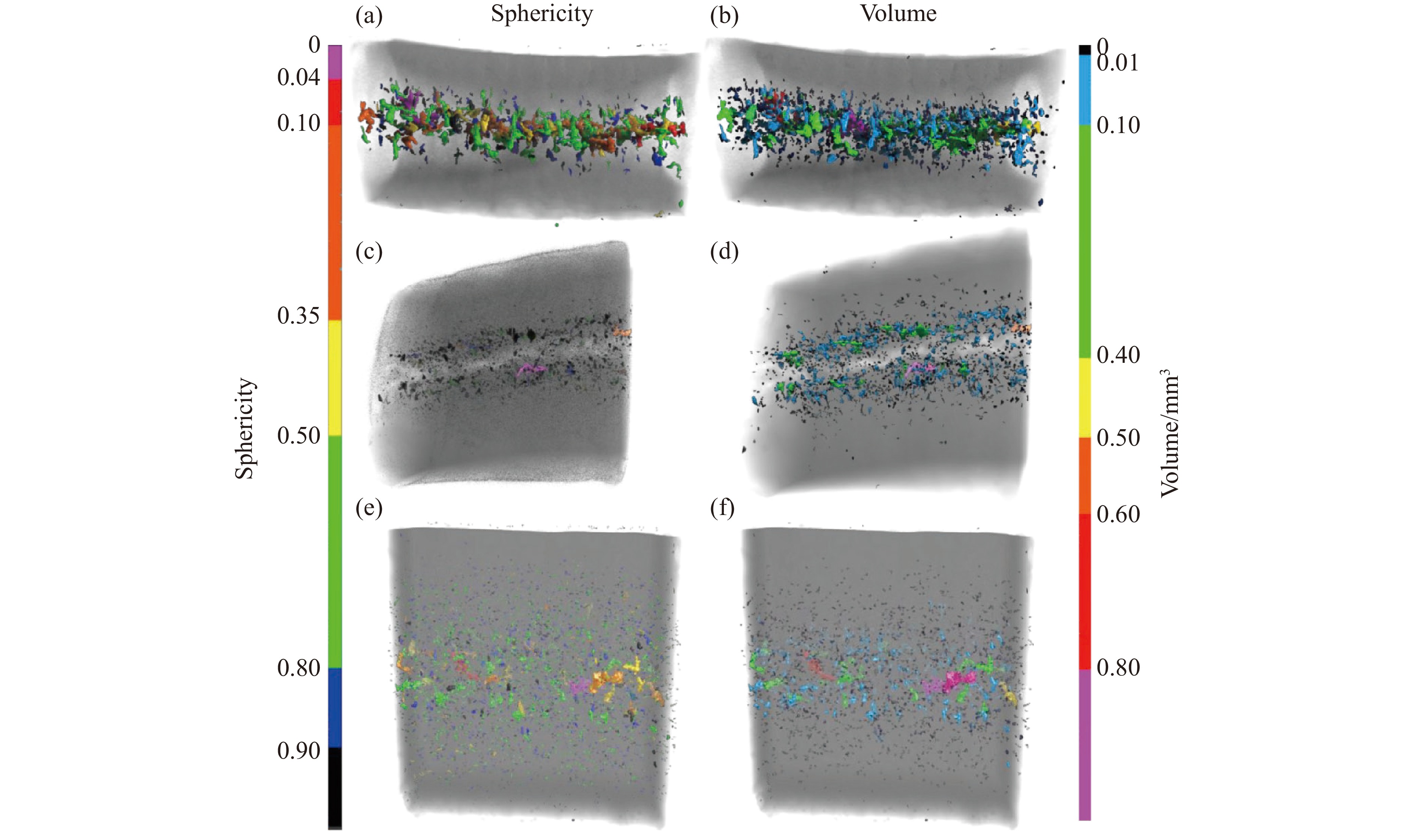
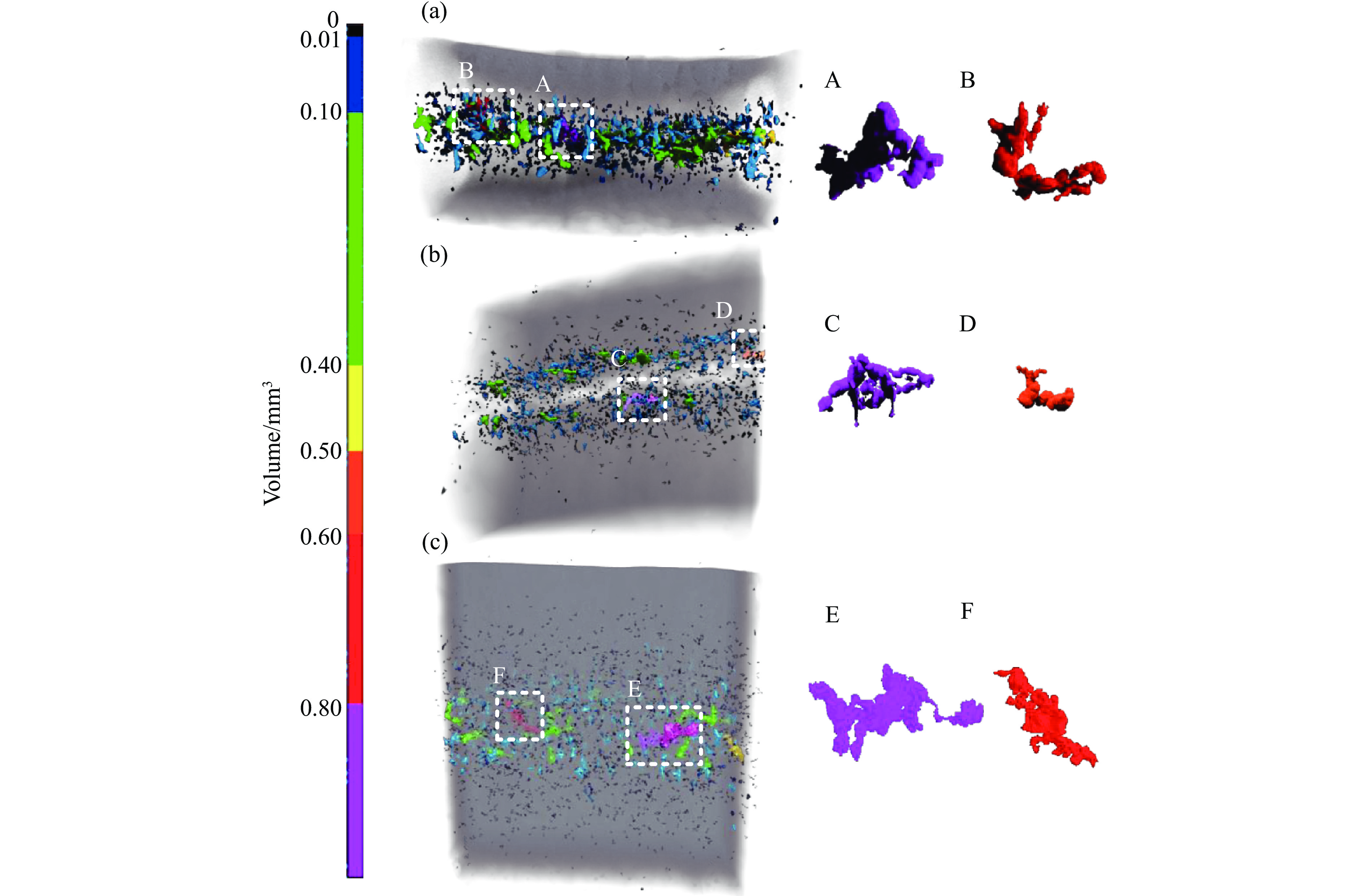
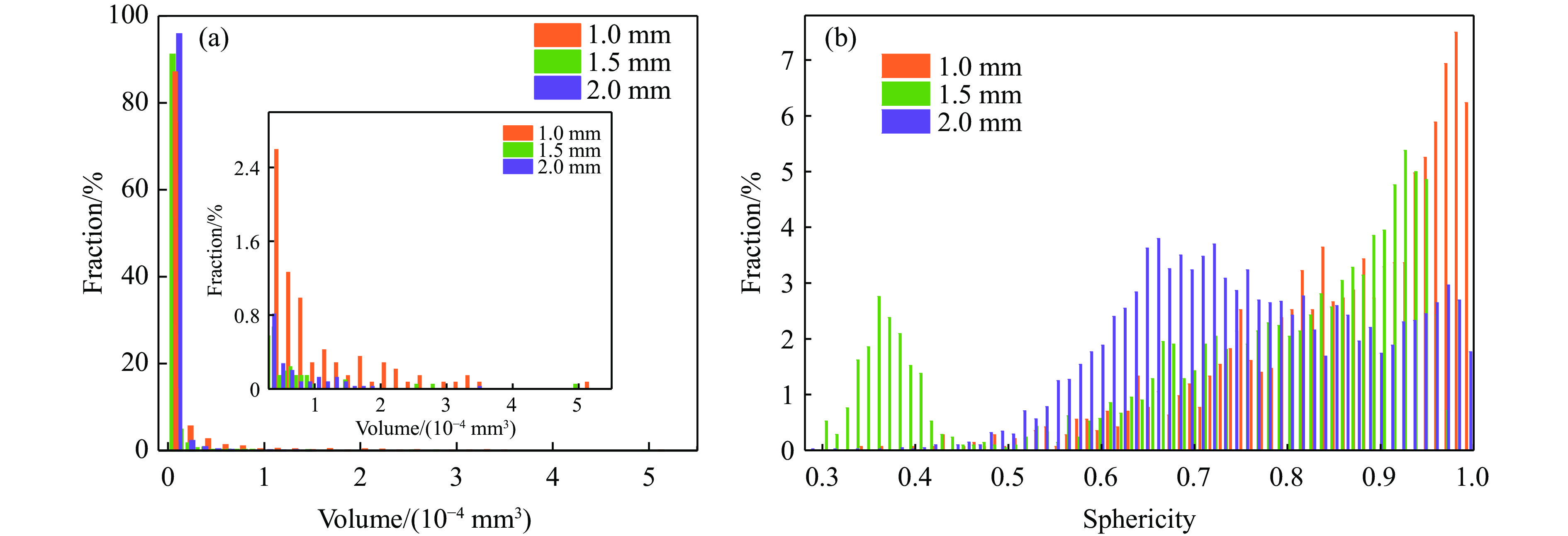
2026,
40(8): 080103.
doi: 10.11858/gywlxb.20261008
Abstract:
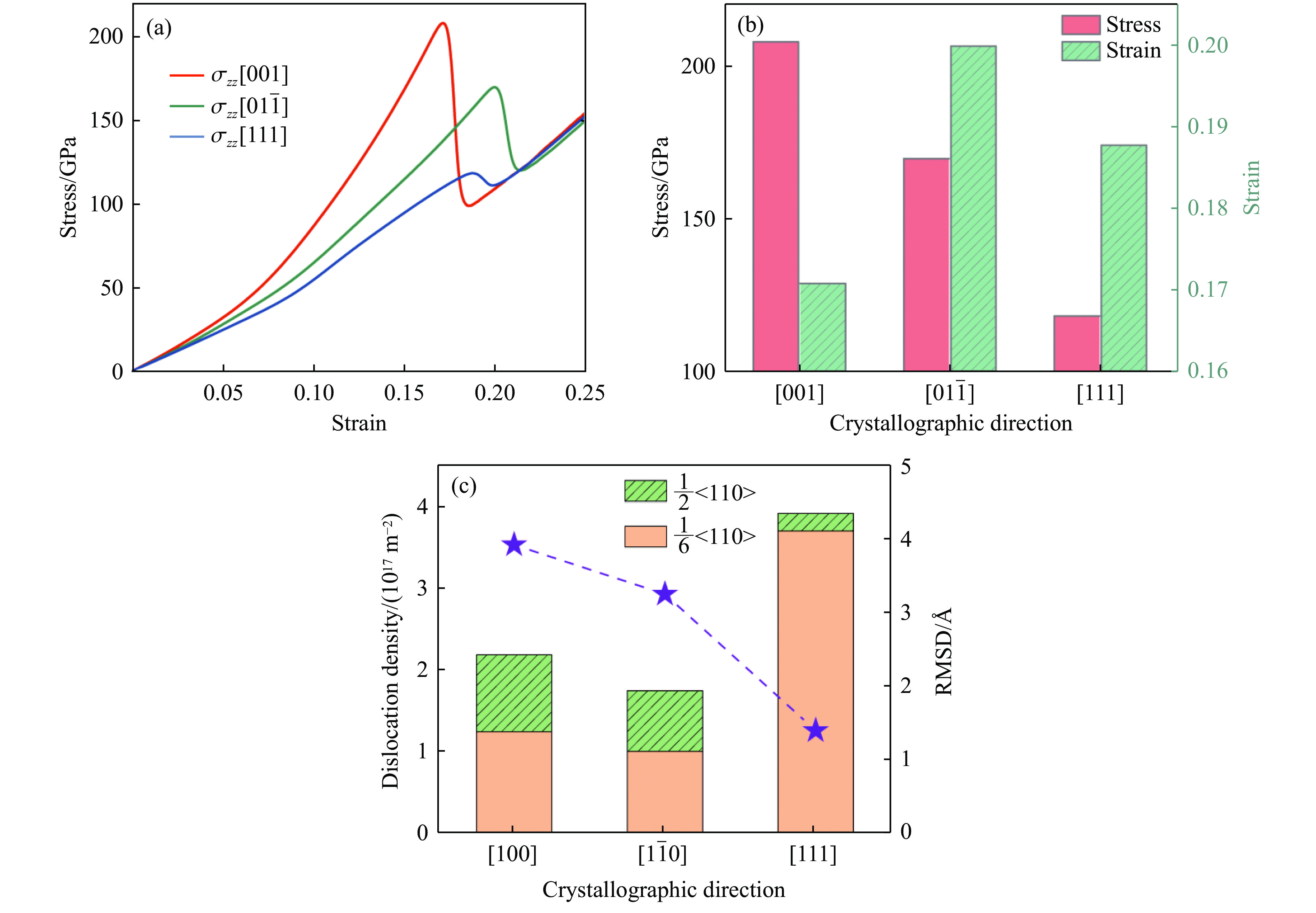
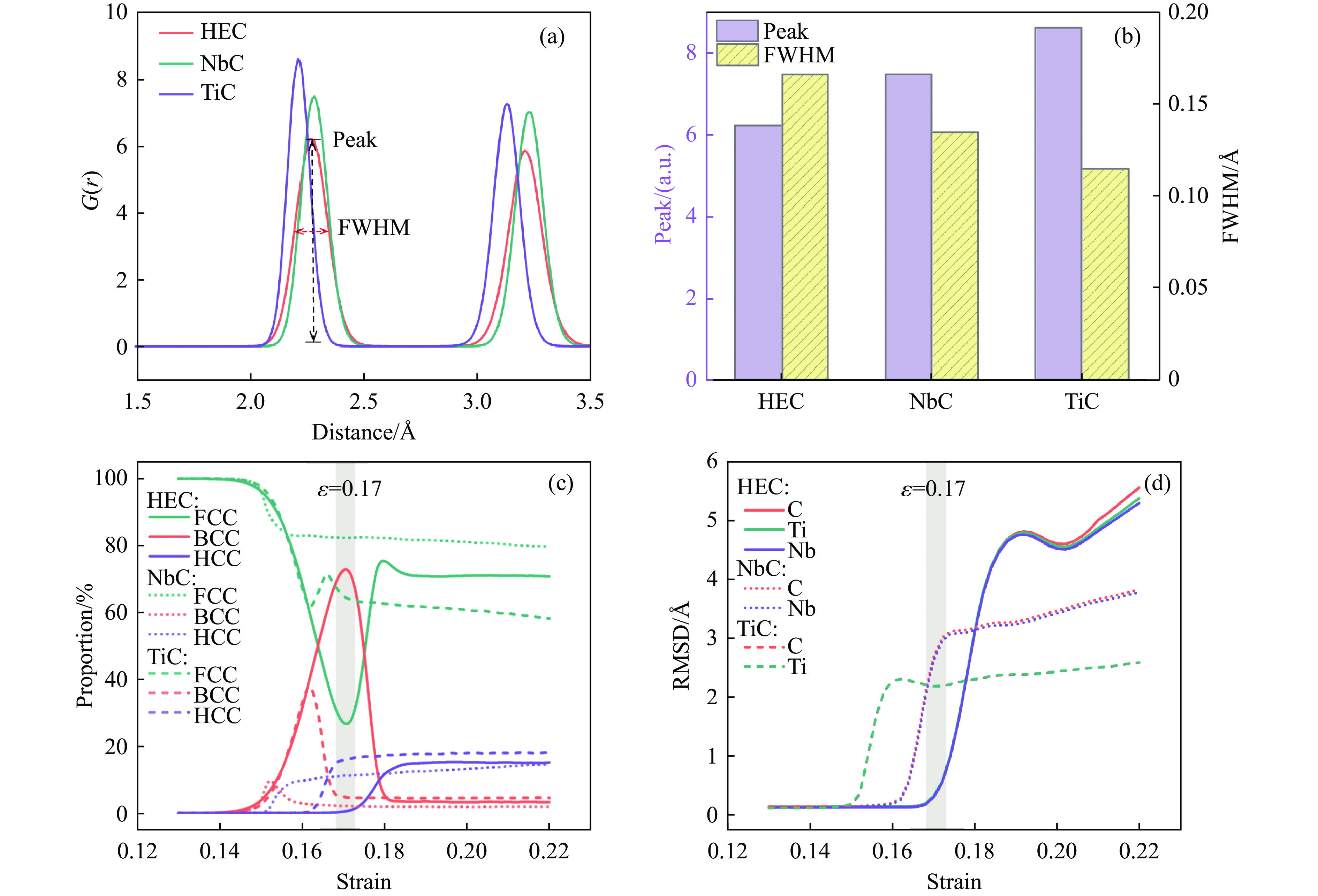
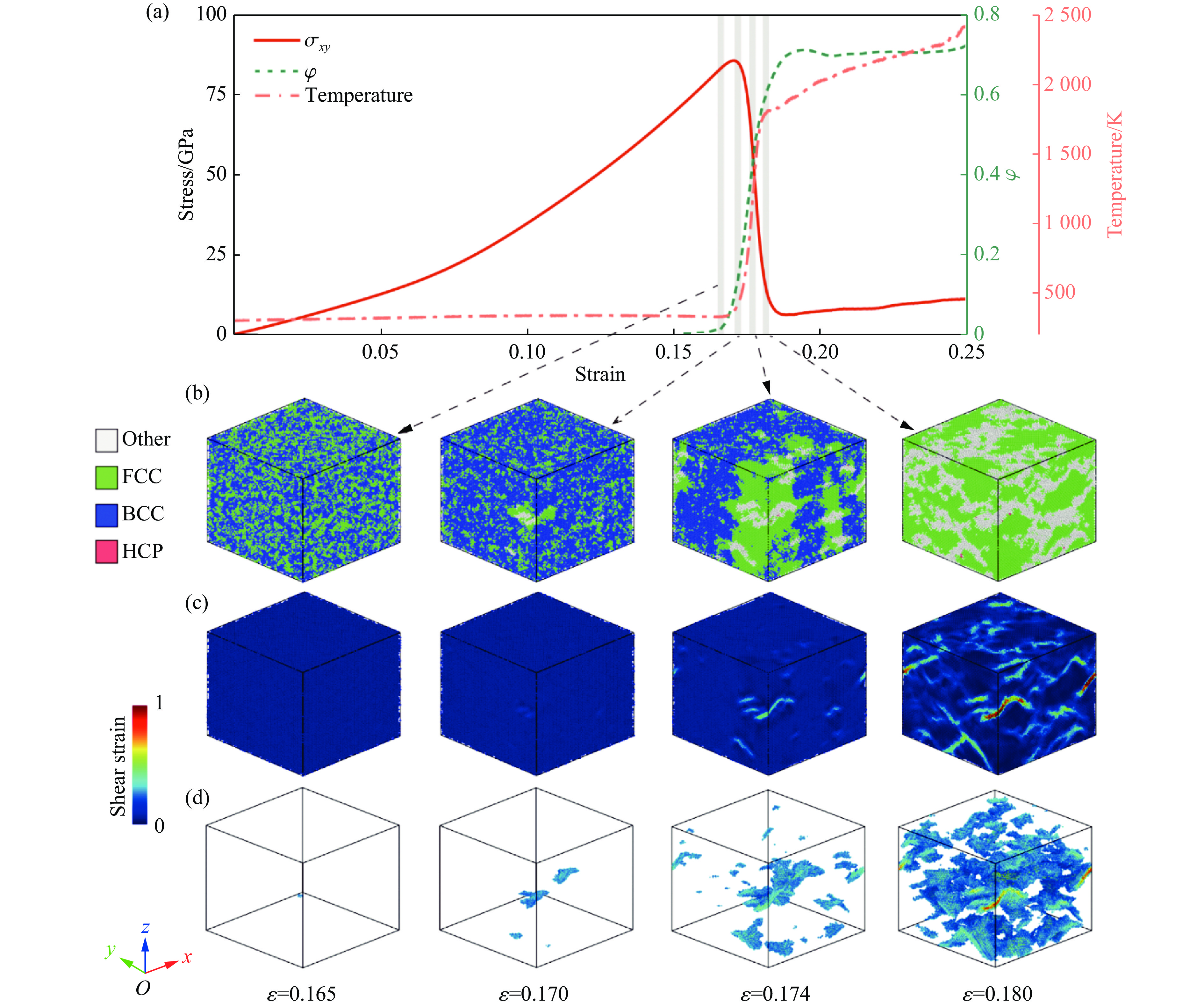
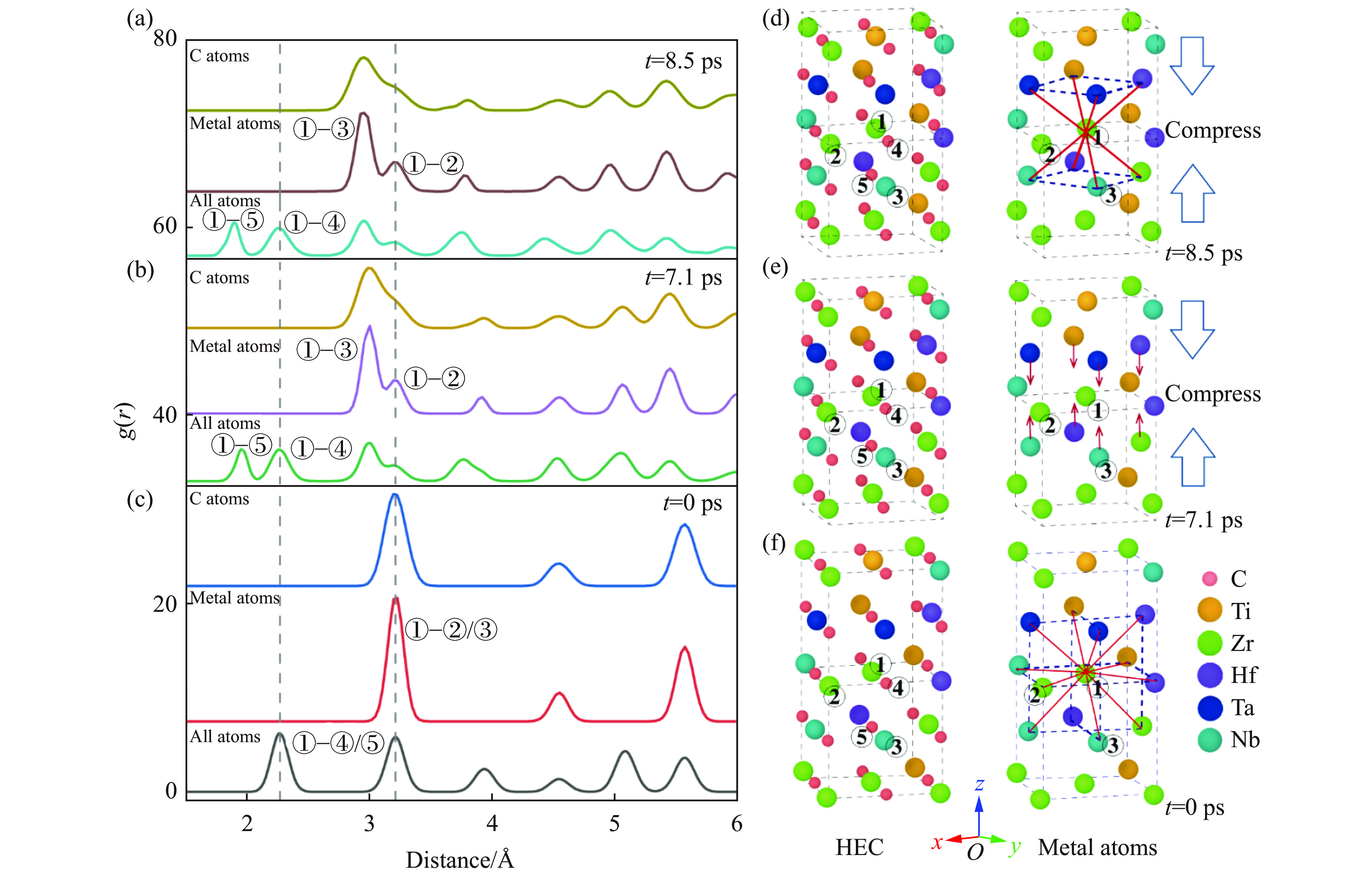
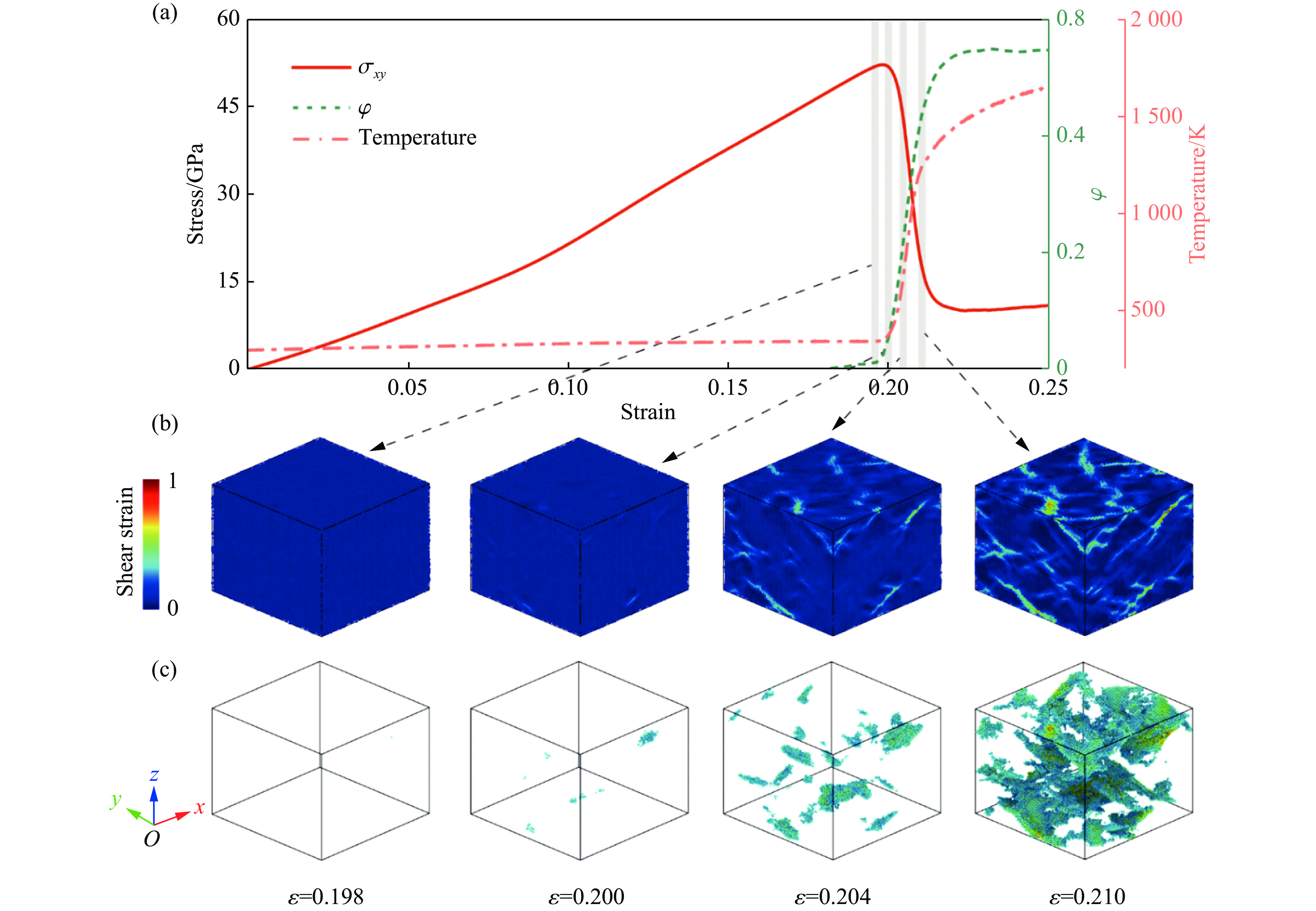
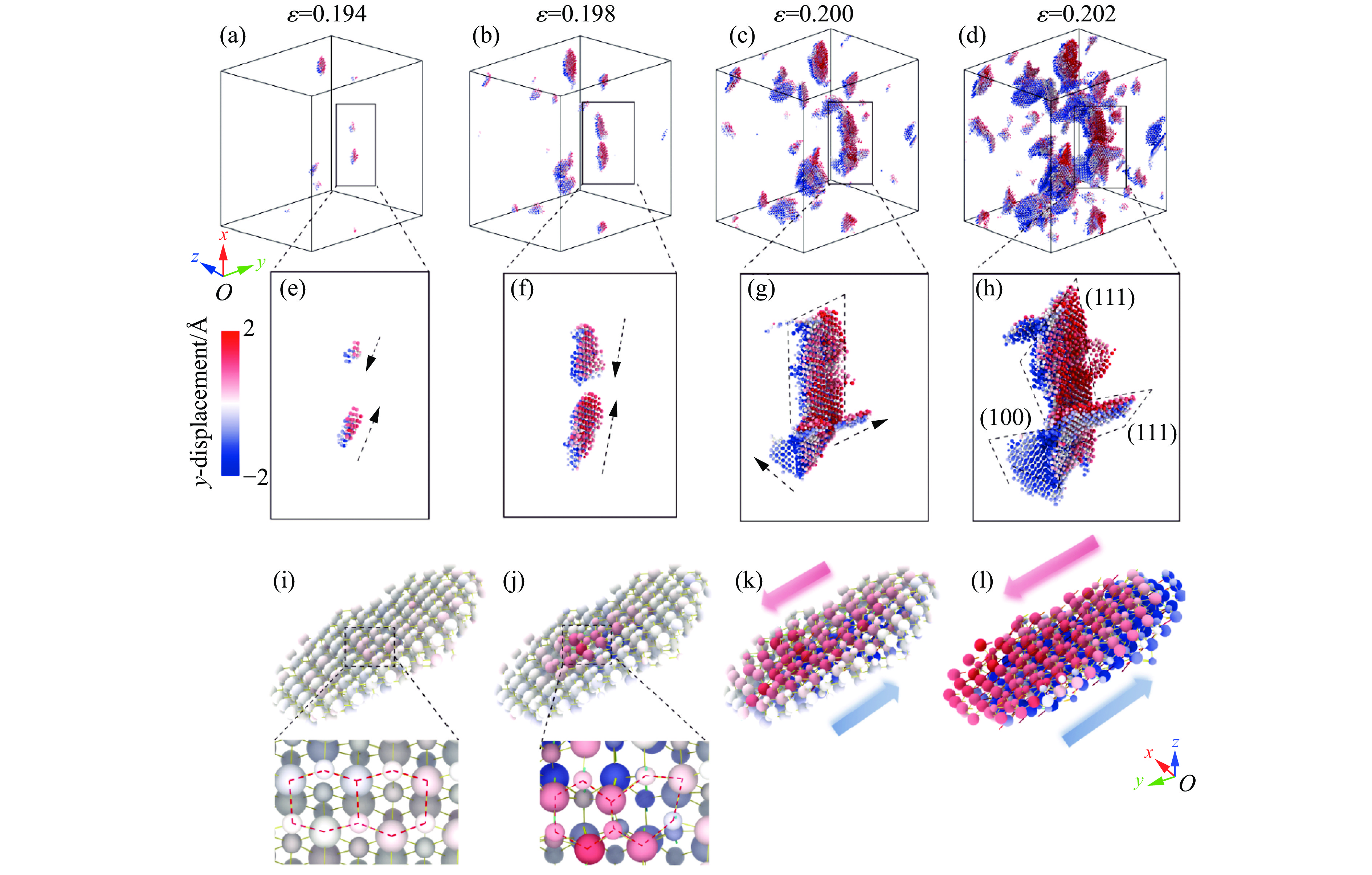
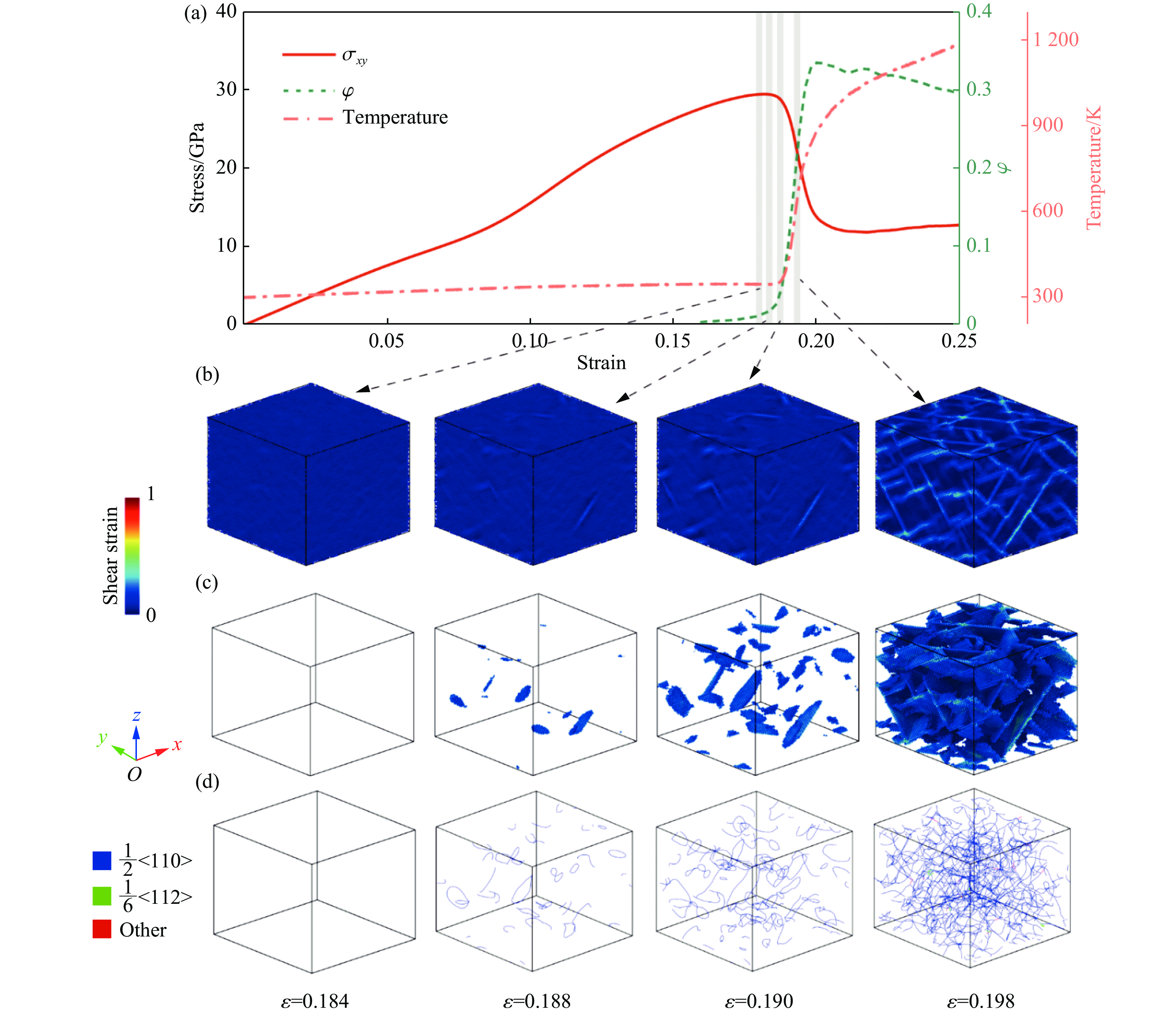

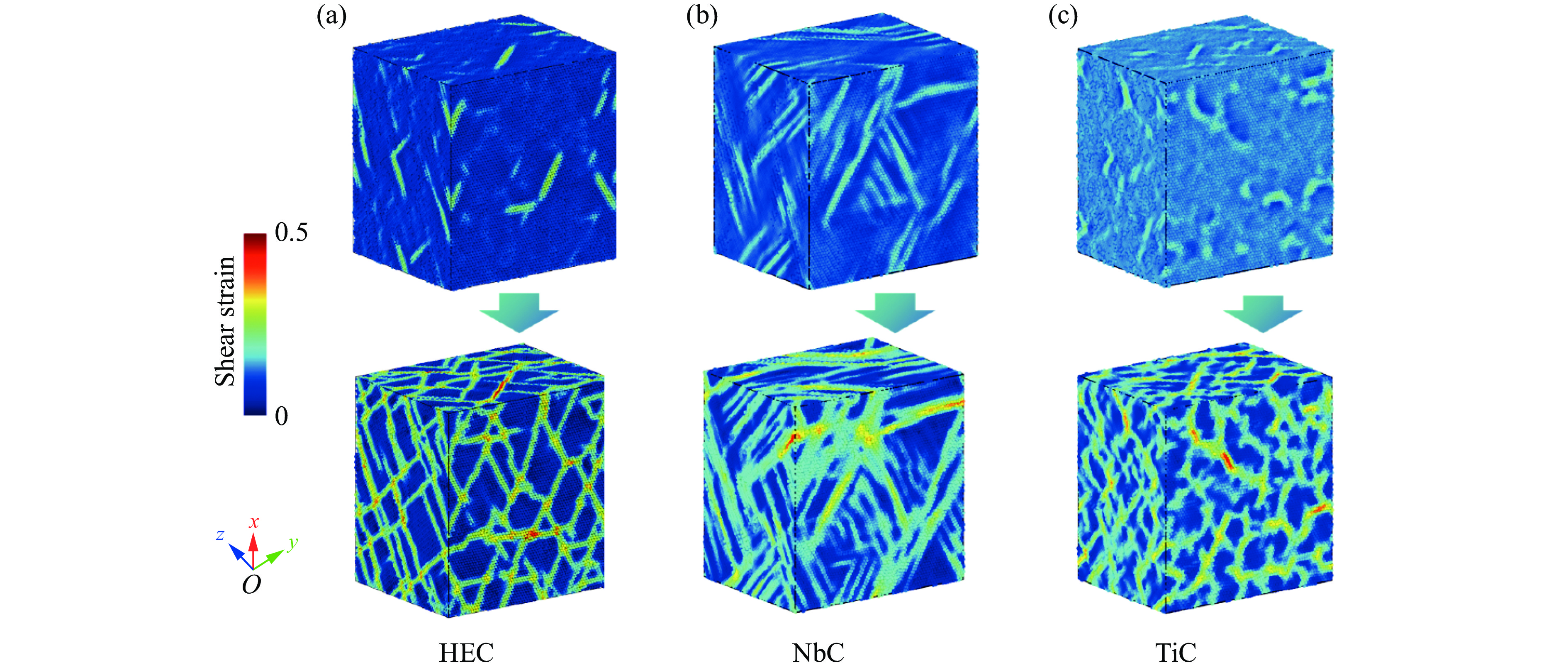
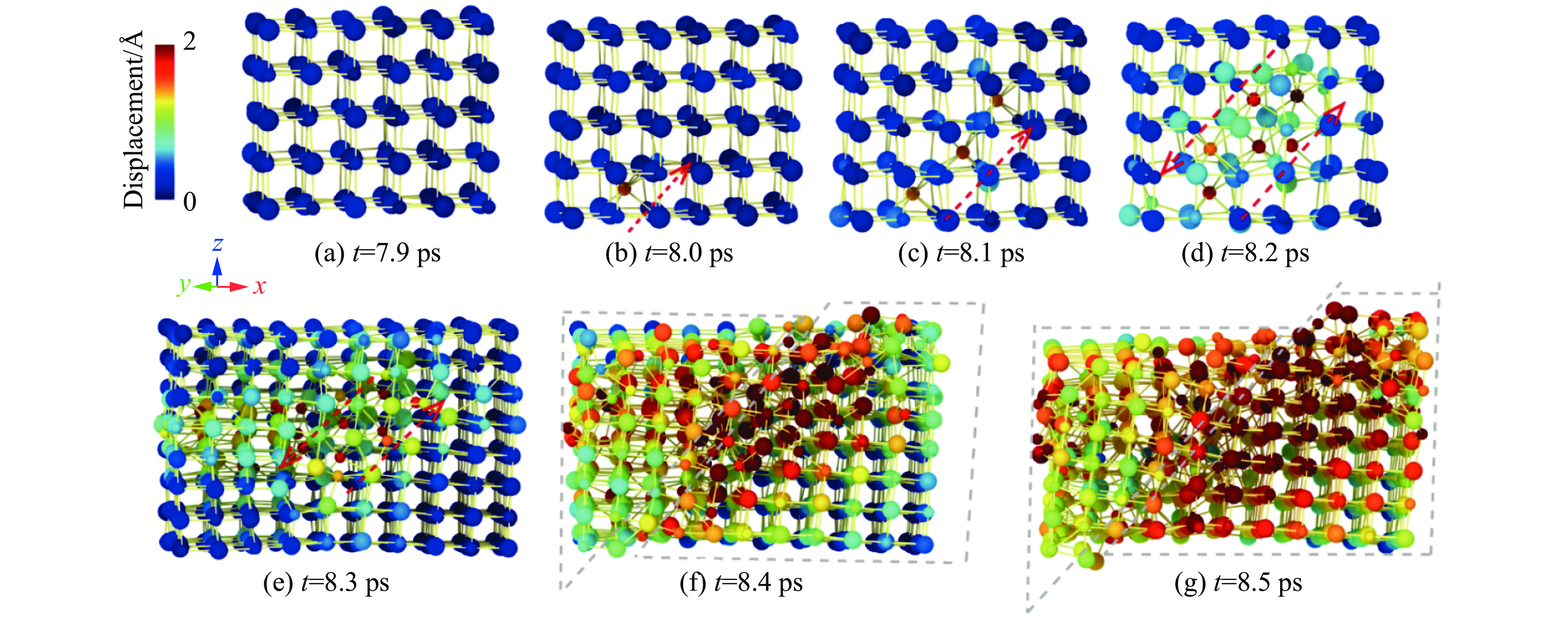
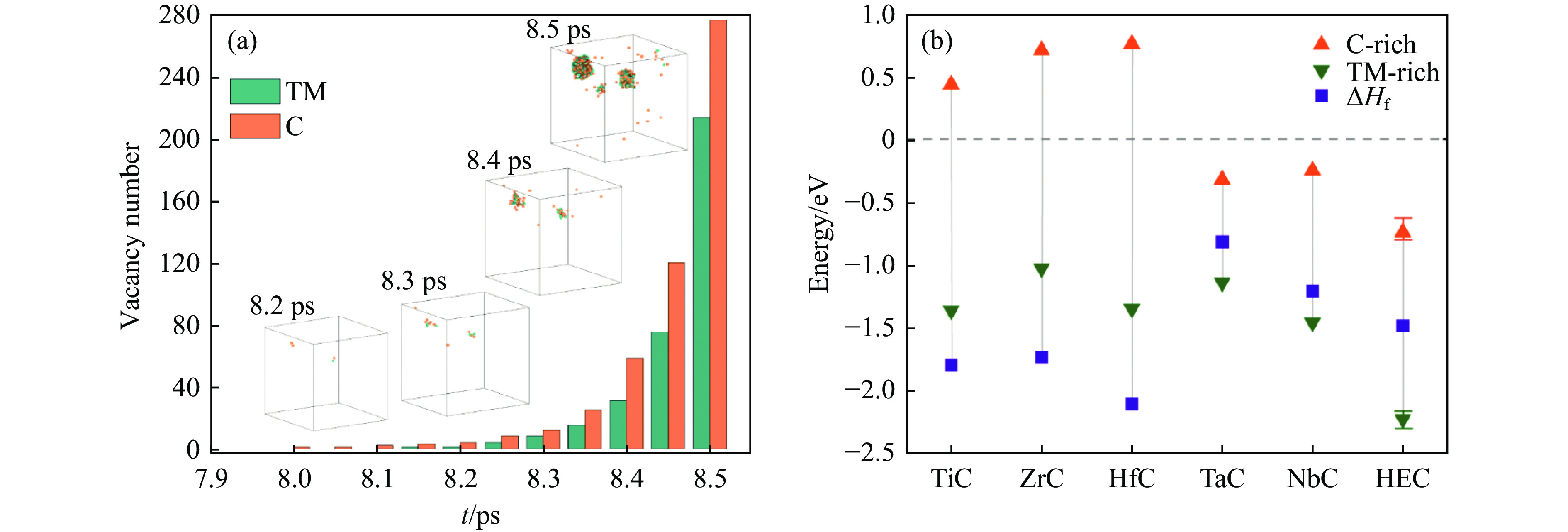
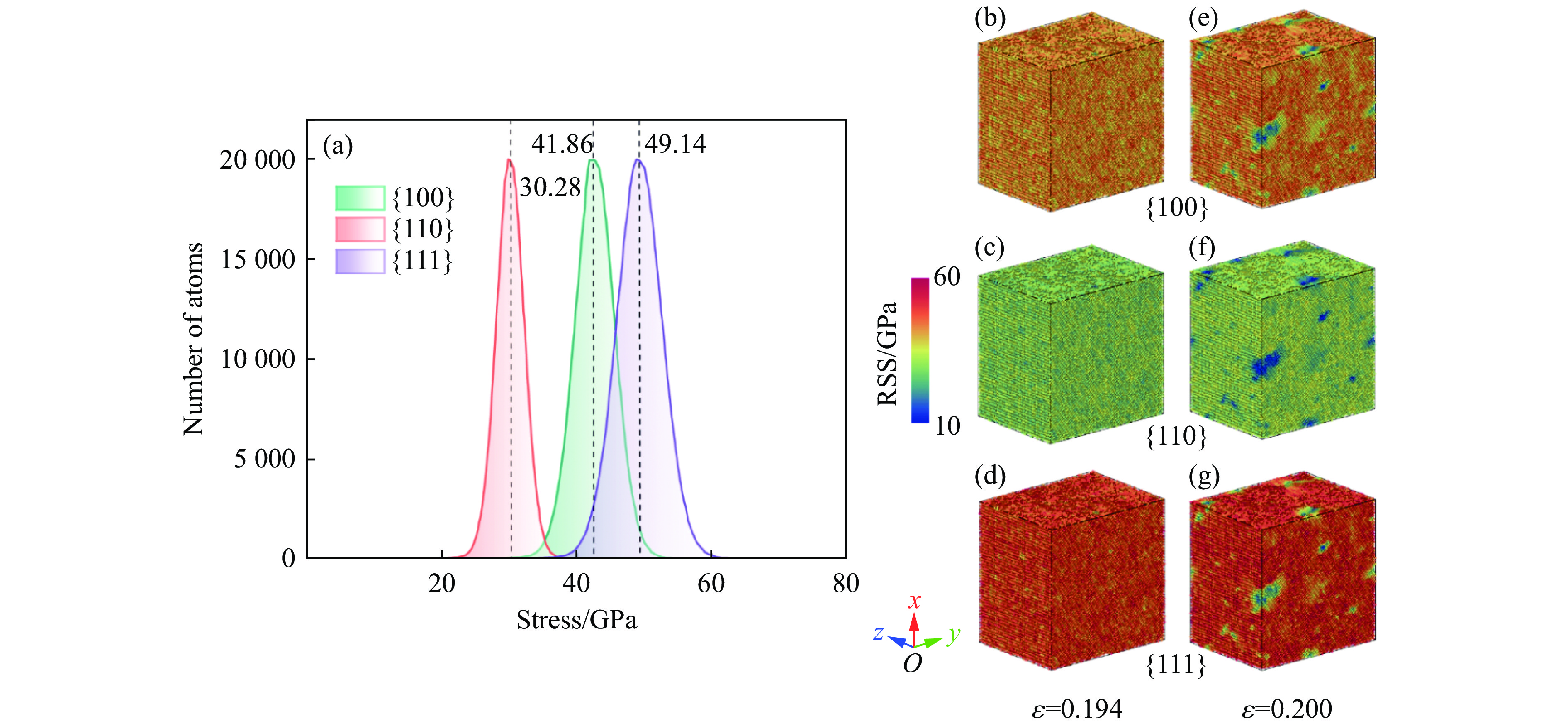
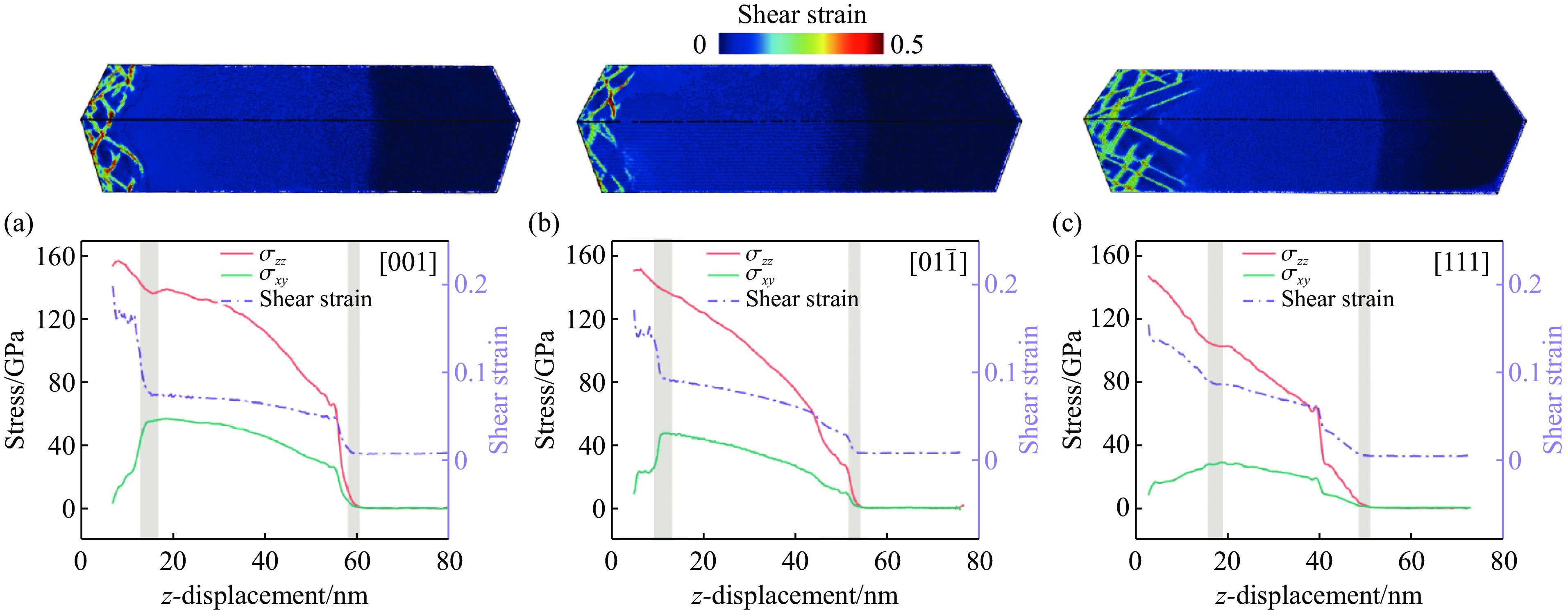
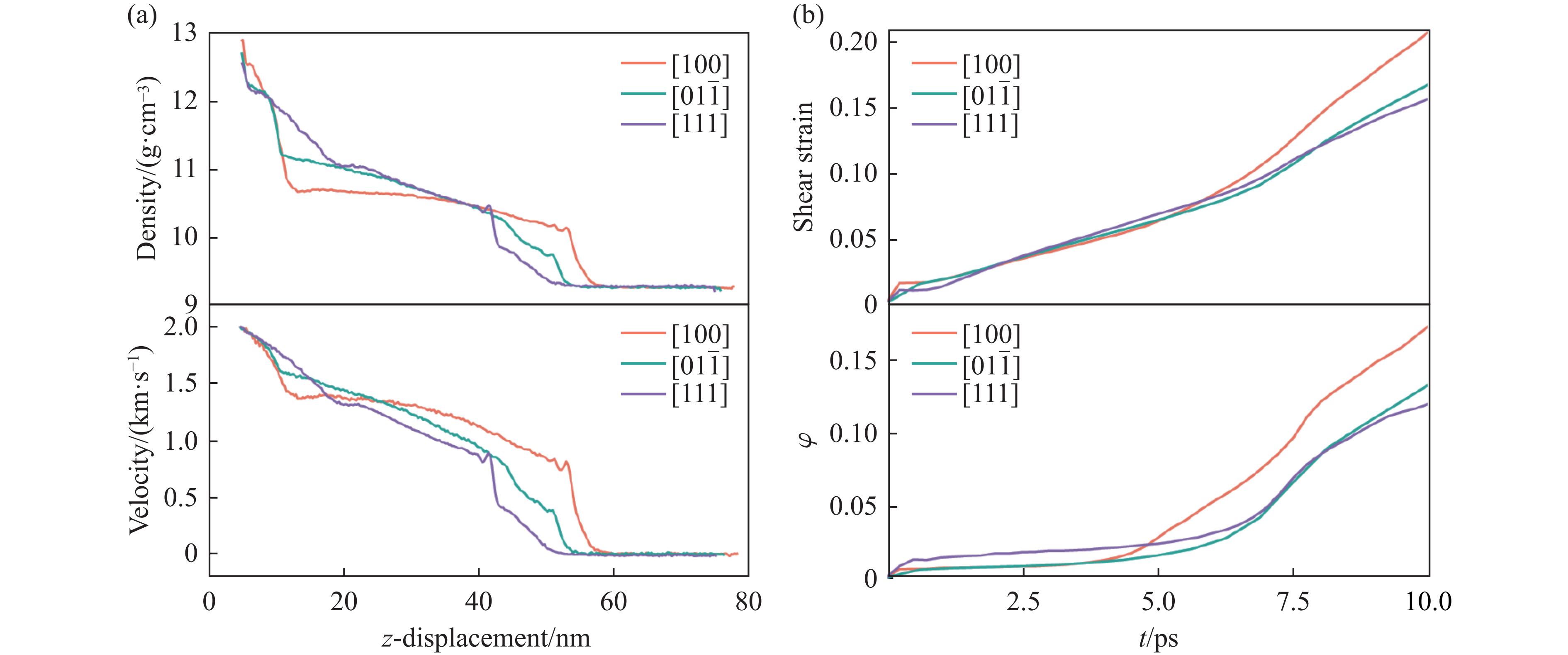
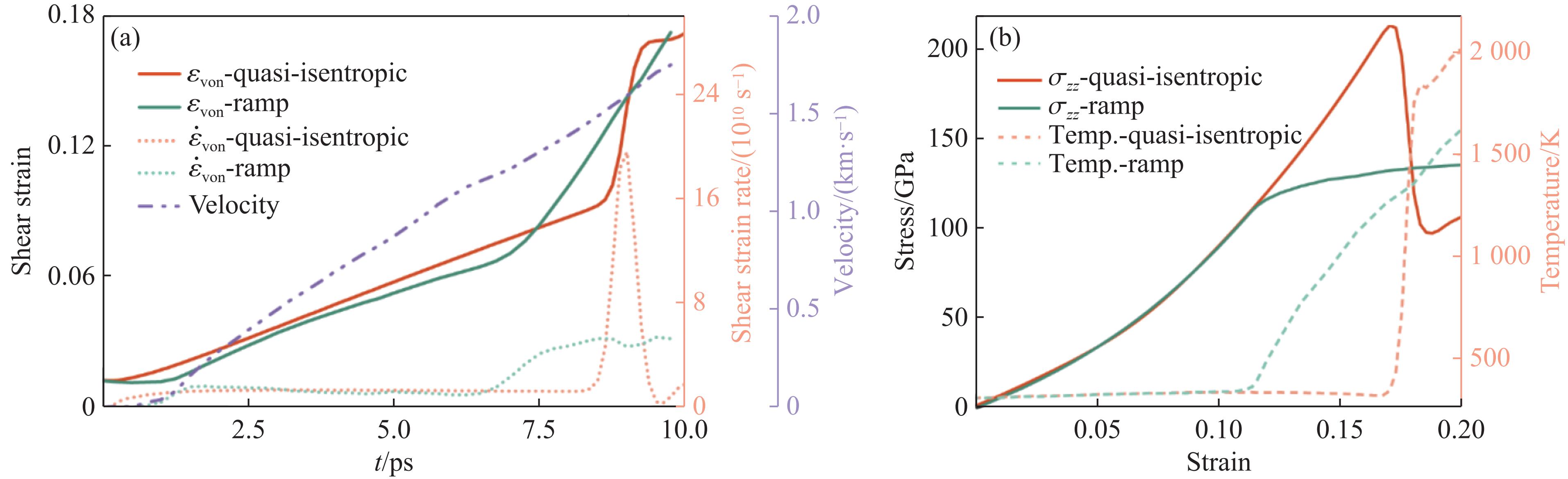
2026,
40(8): 080104.
doi: 10.11858/gywlxb.20251075
Abstract:
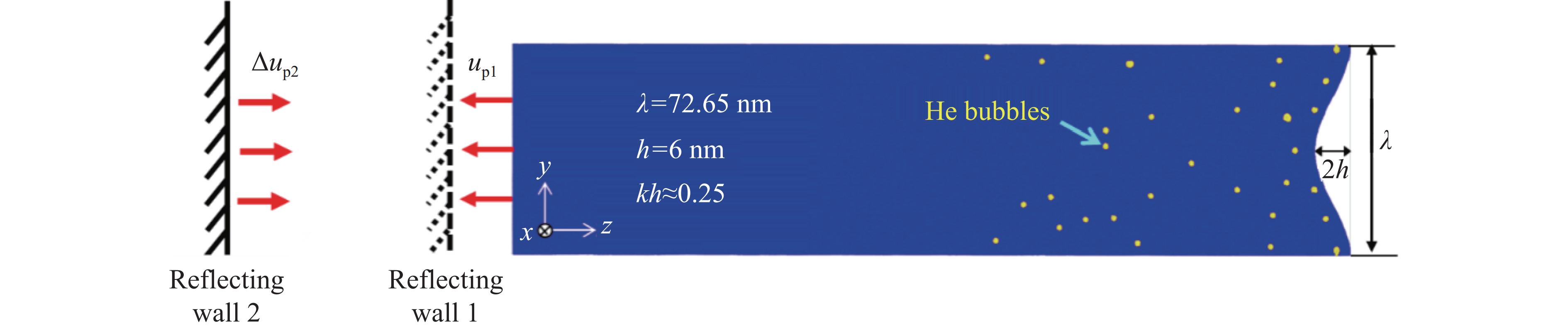
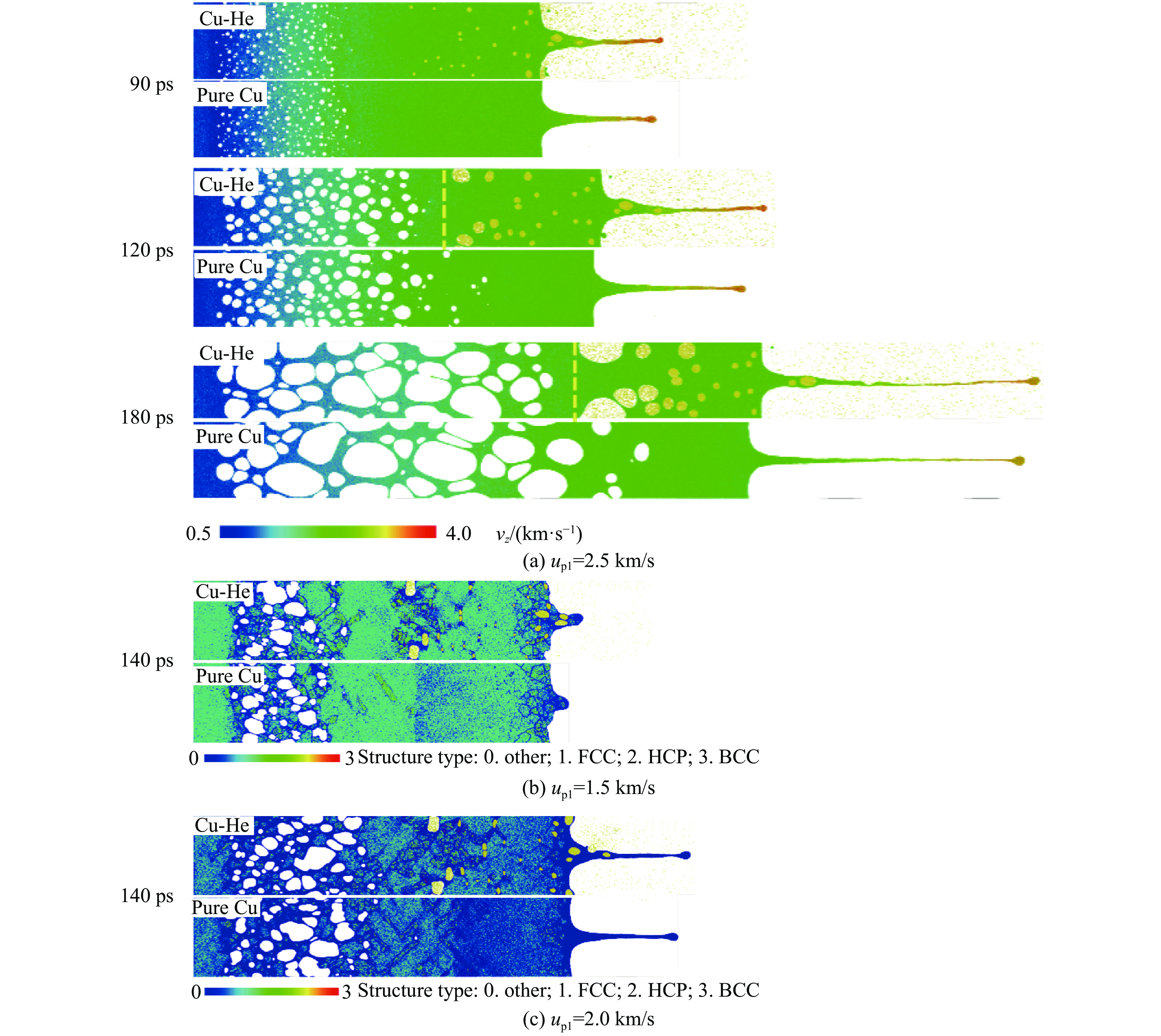
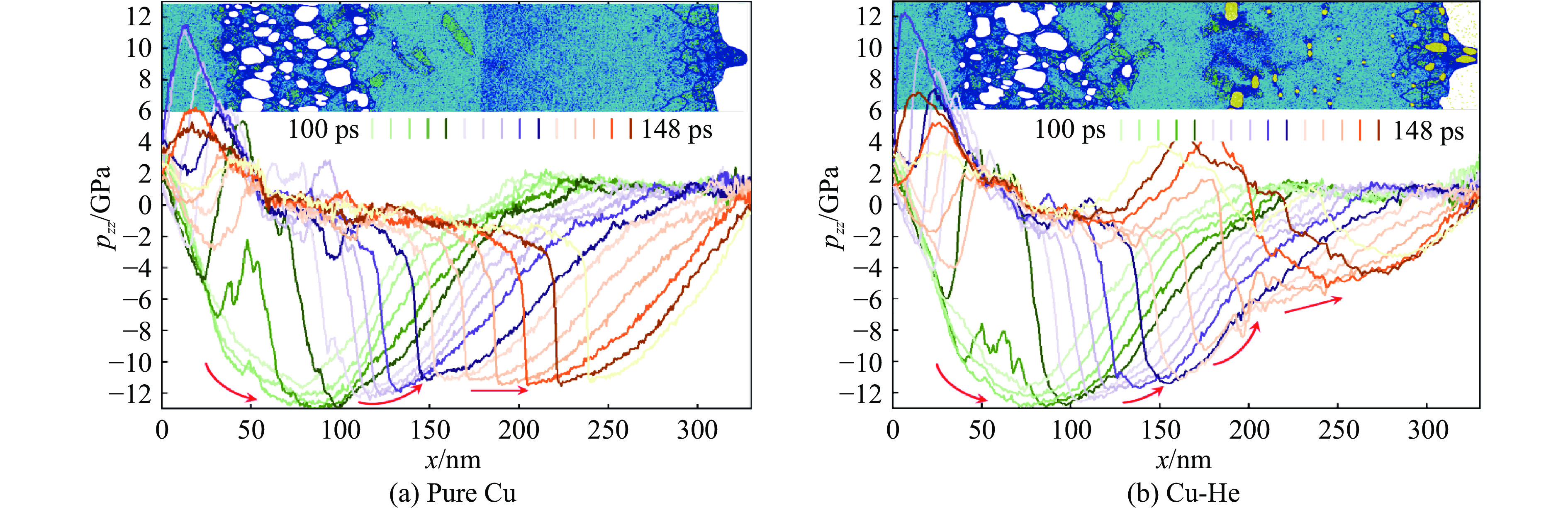
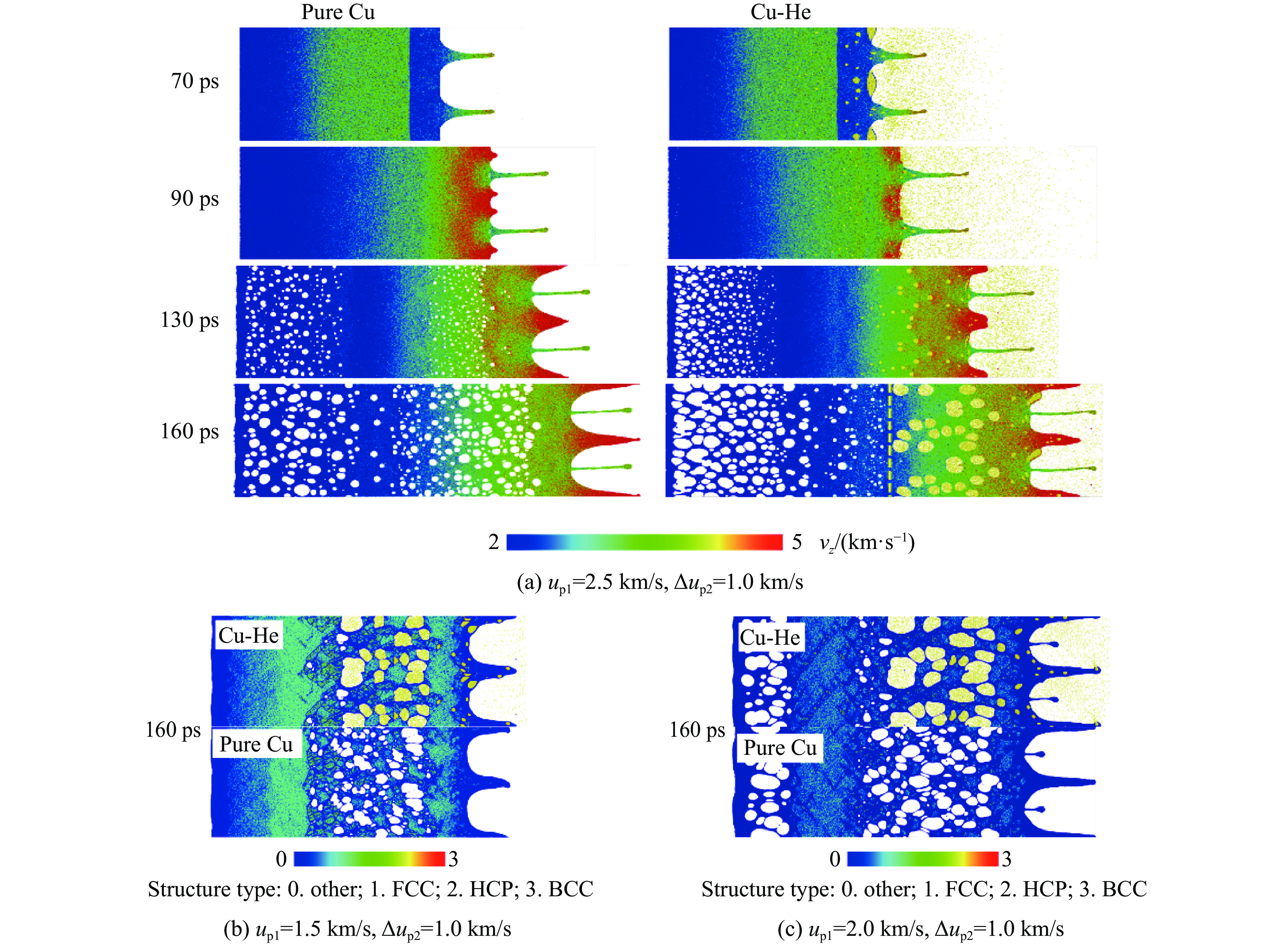
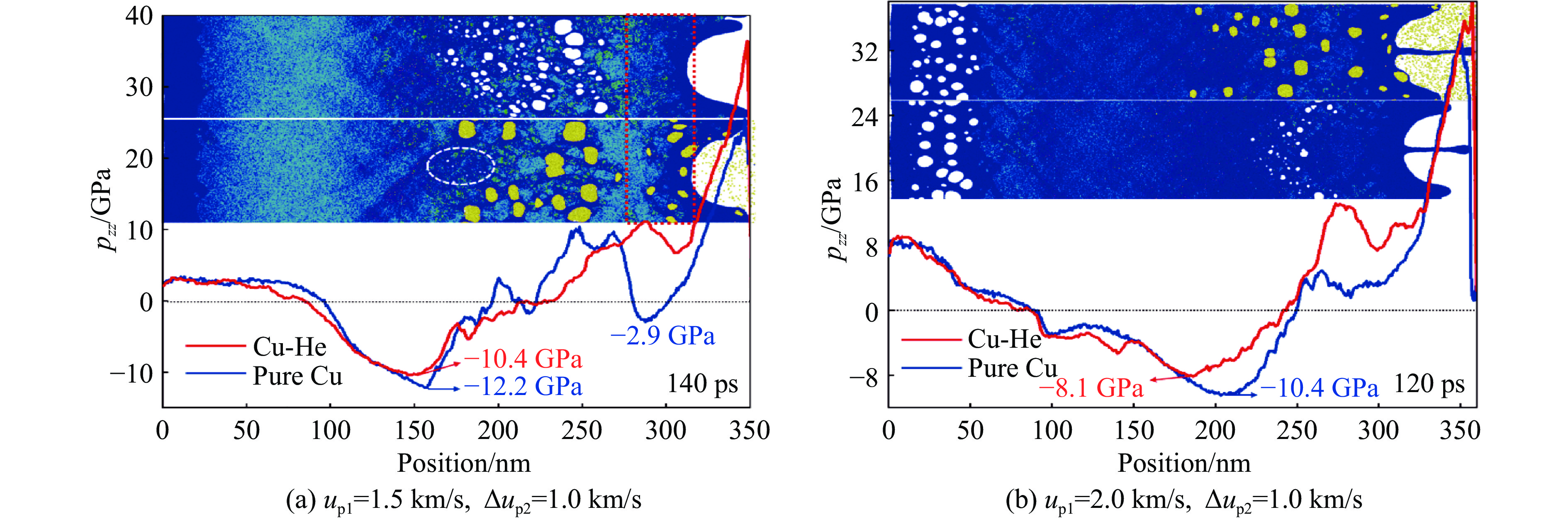
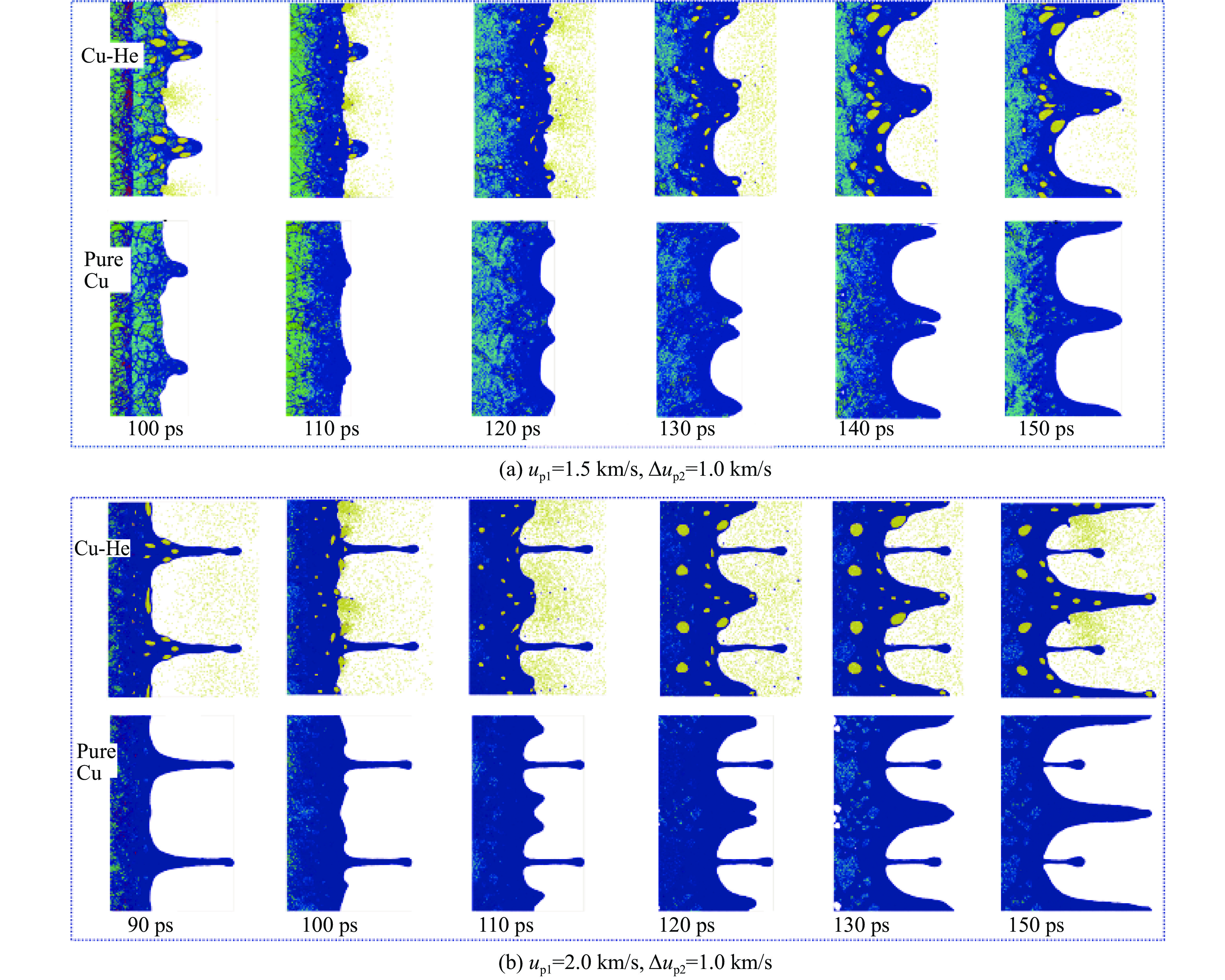
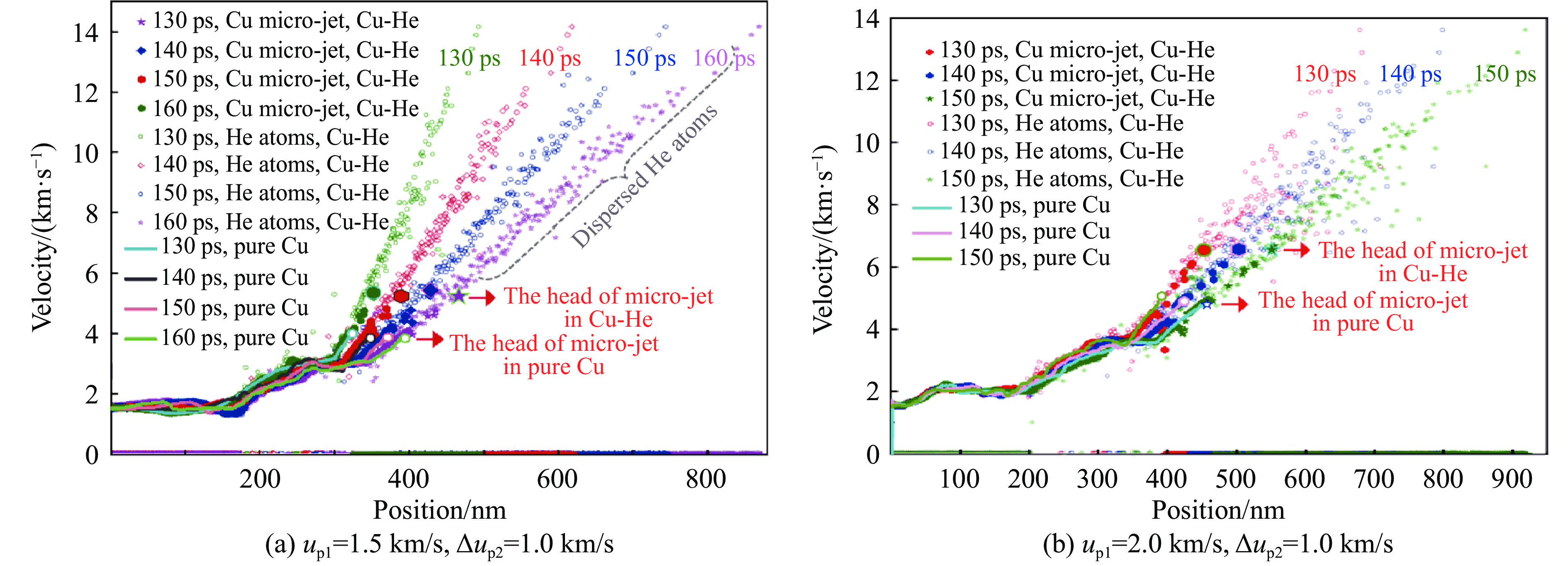
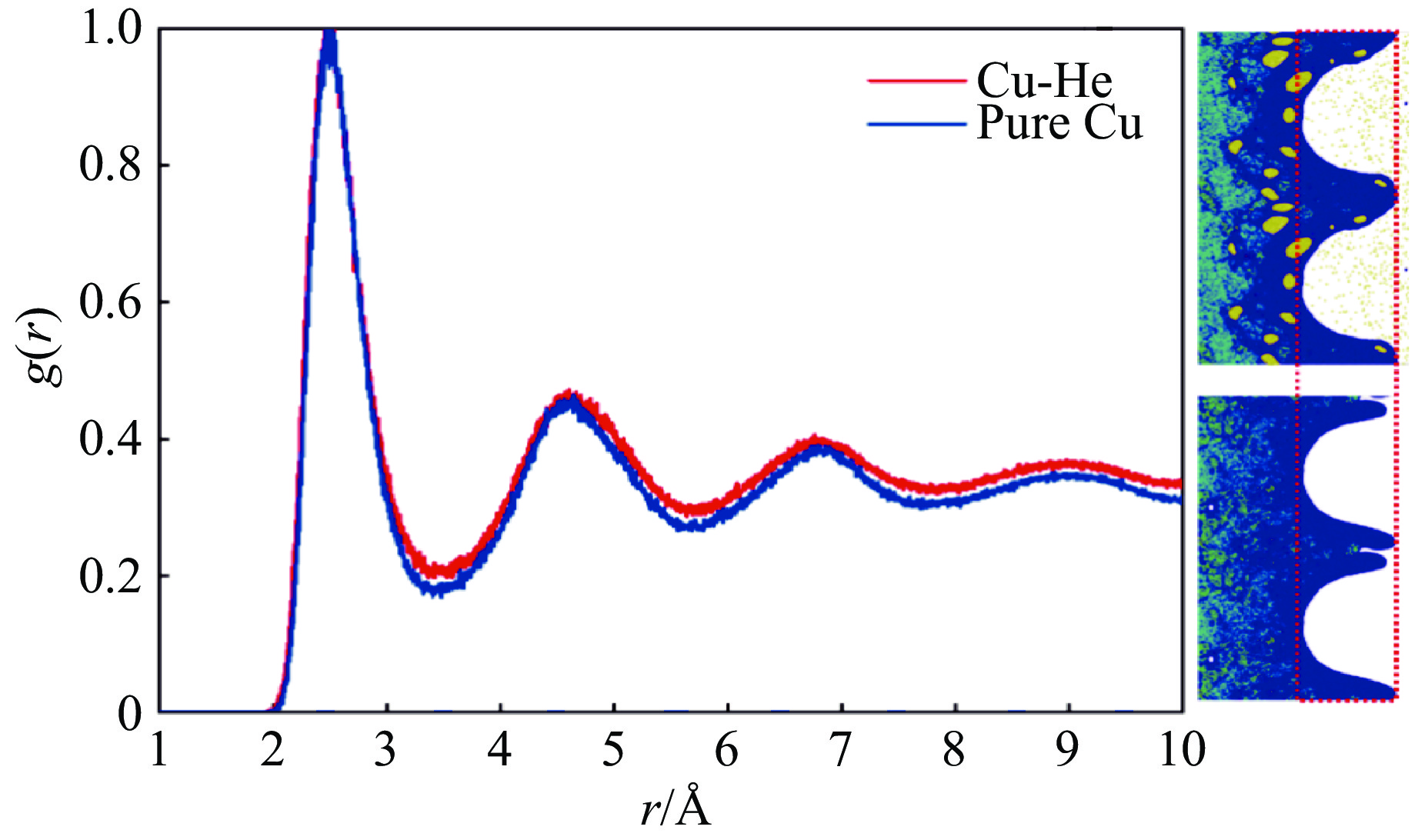
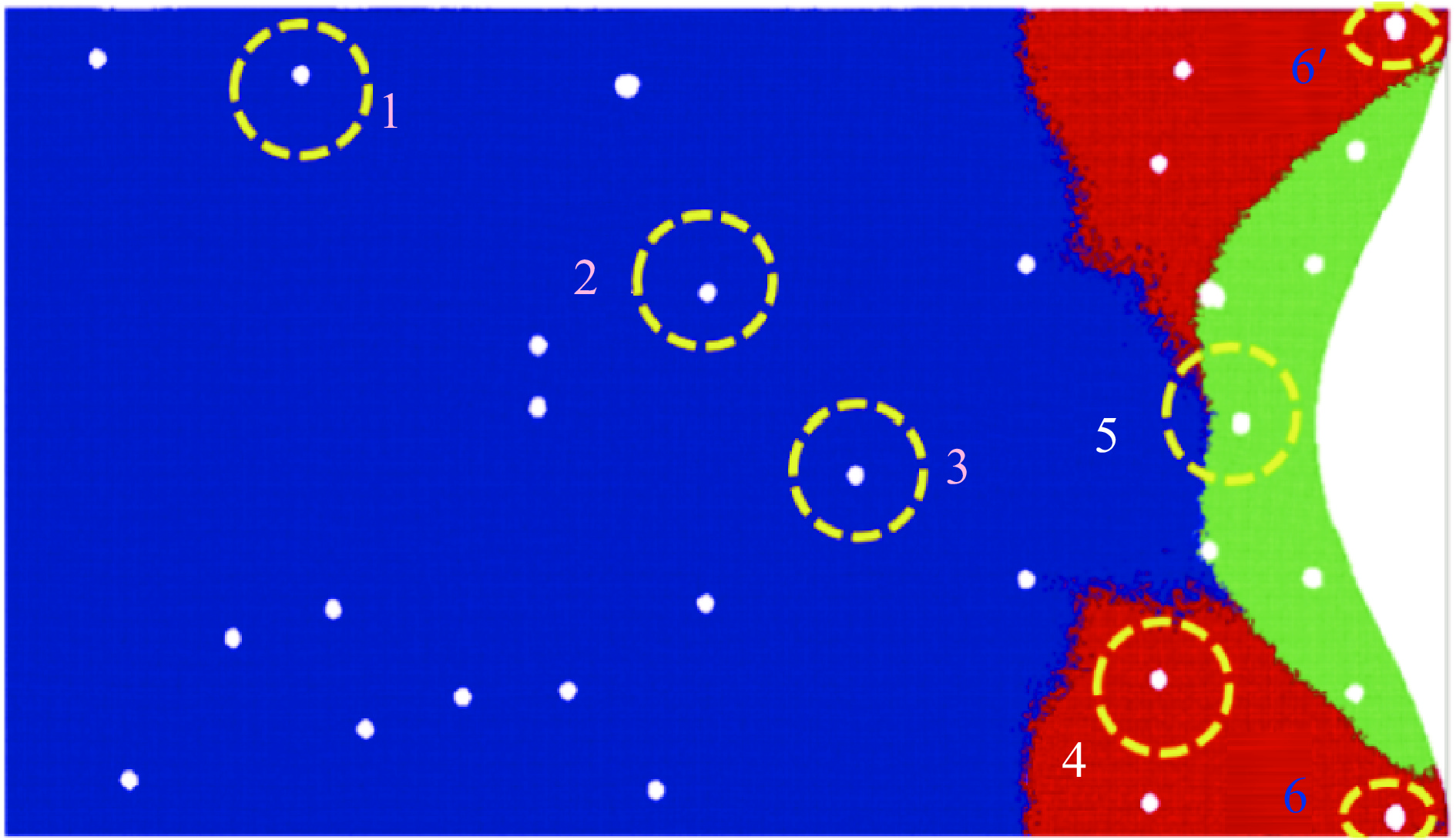
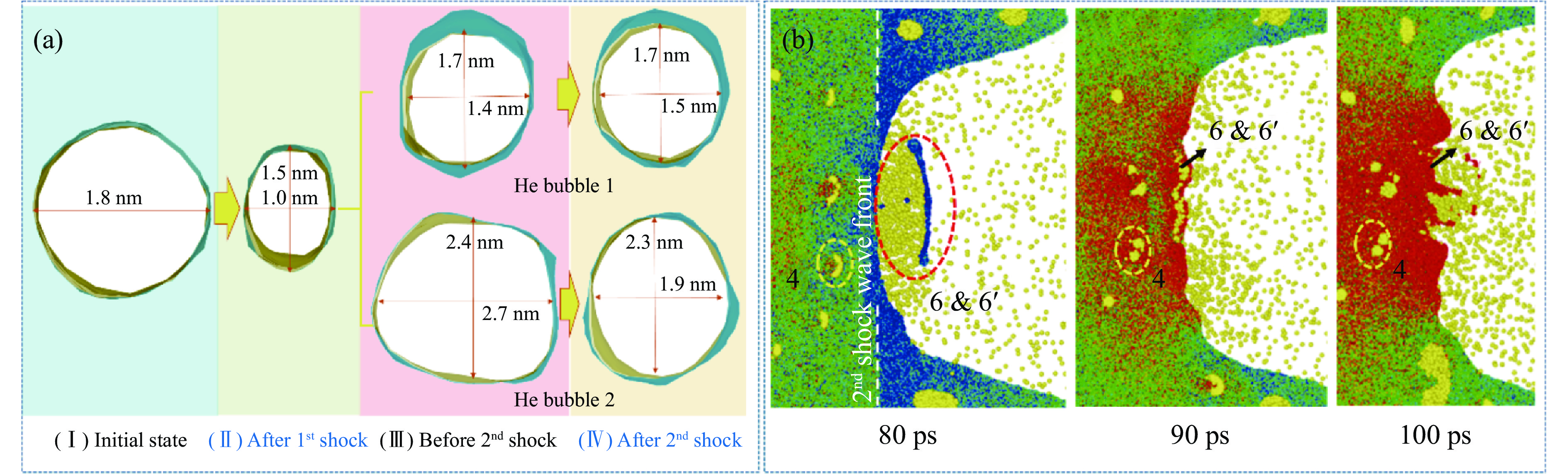
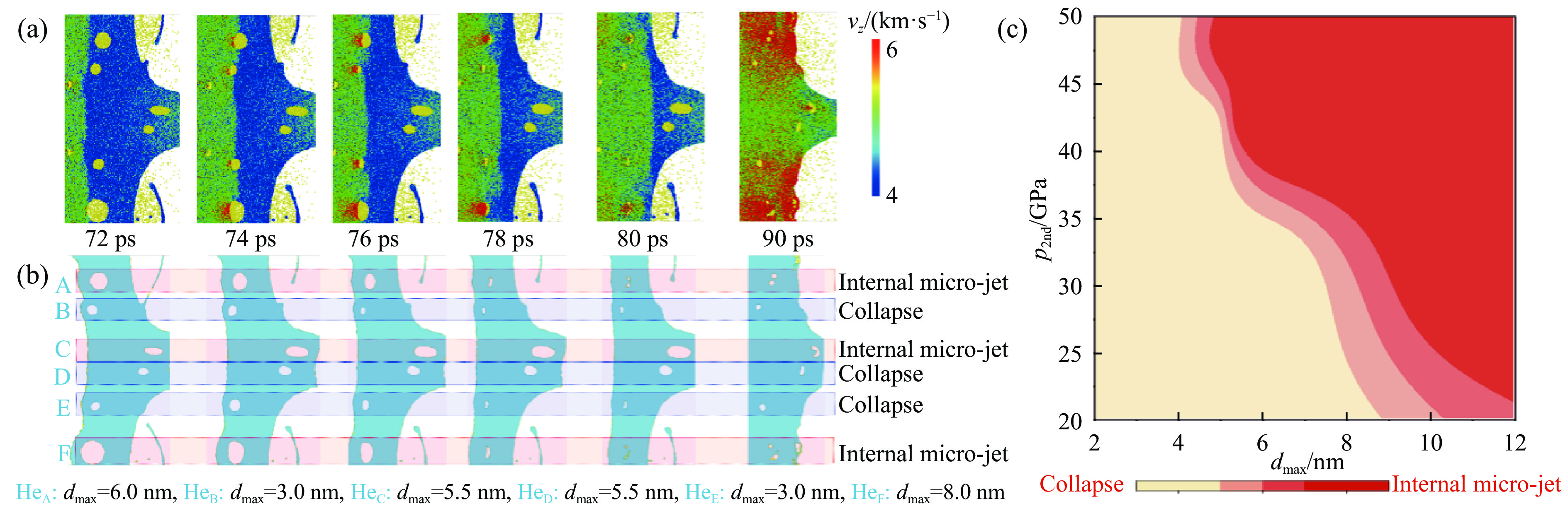
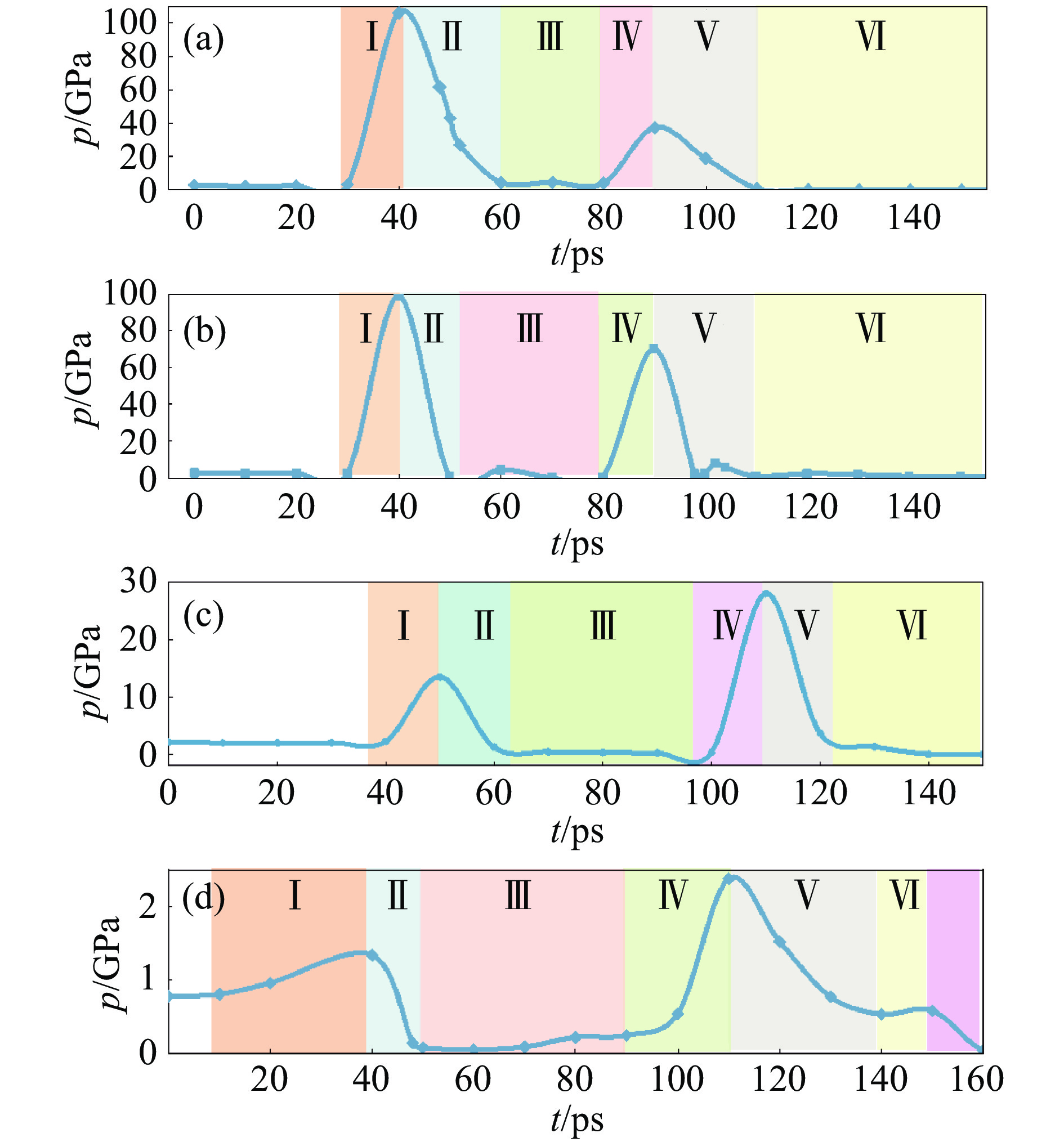
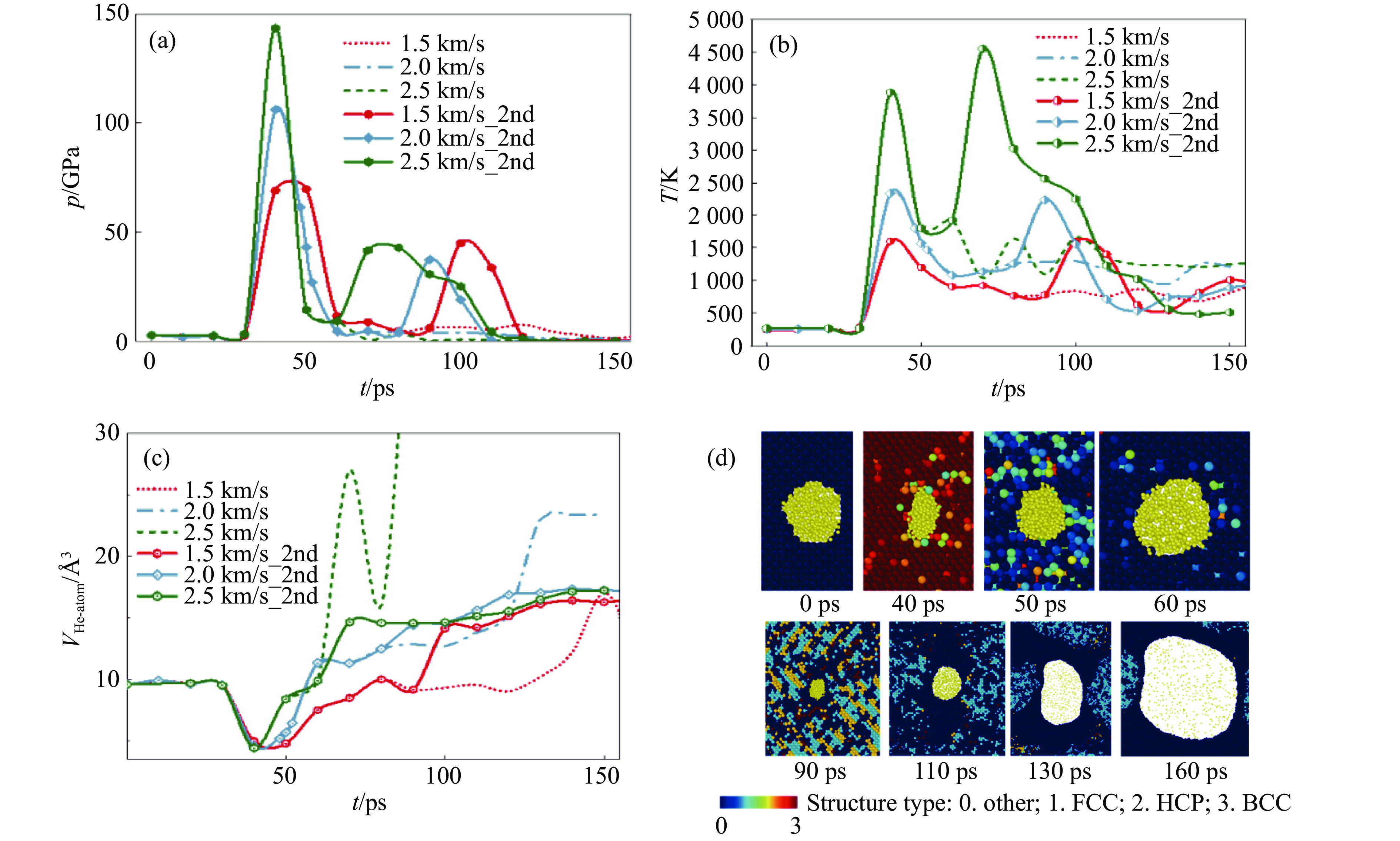
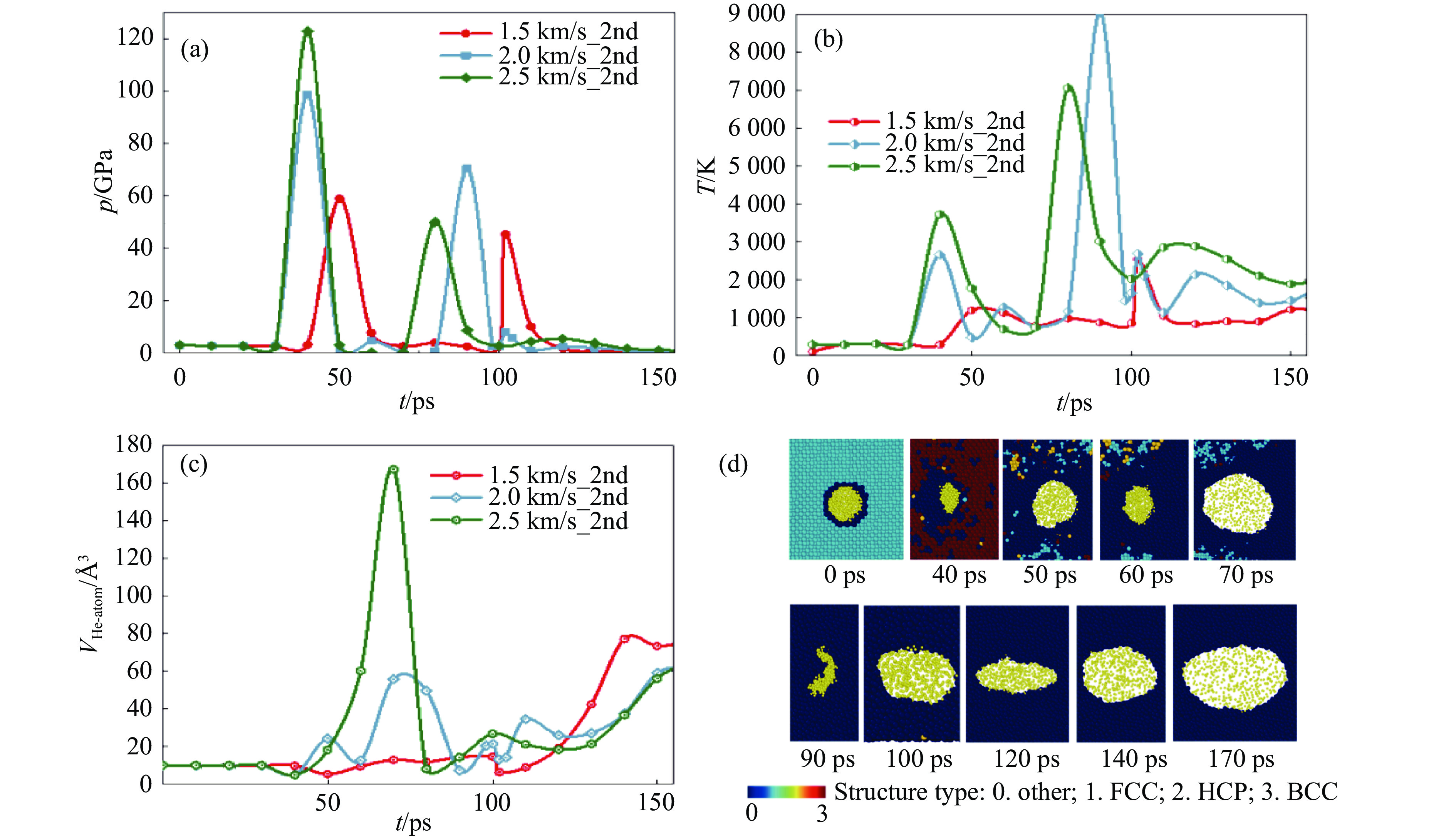
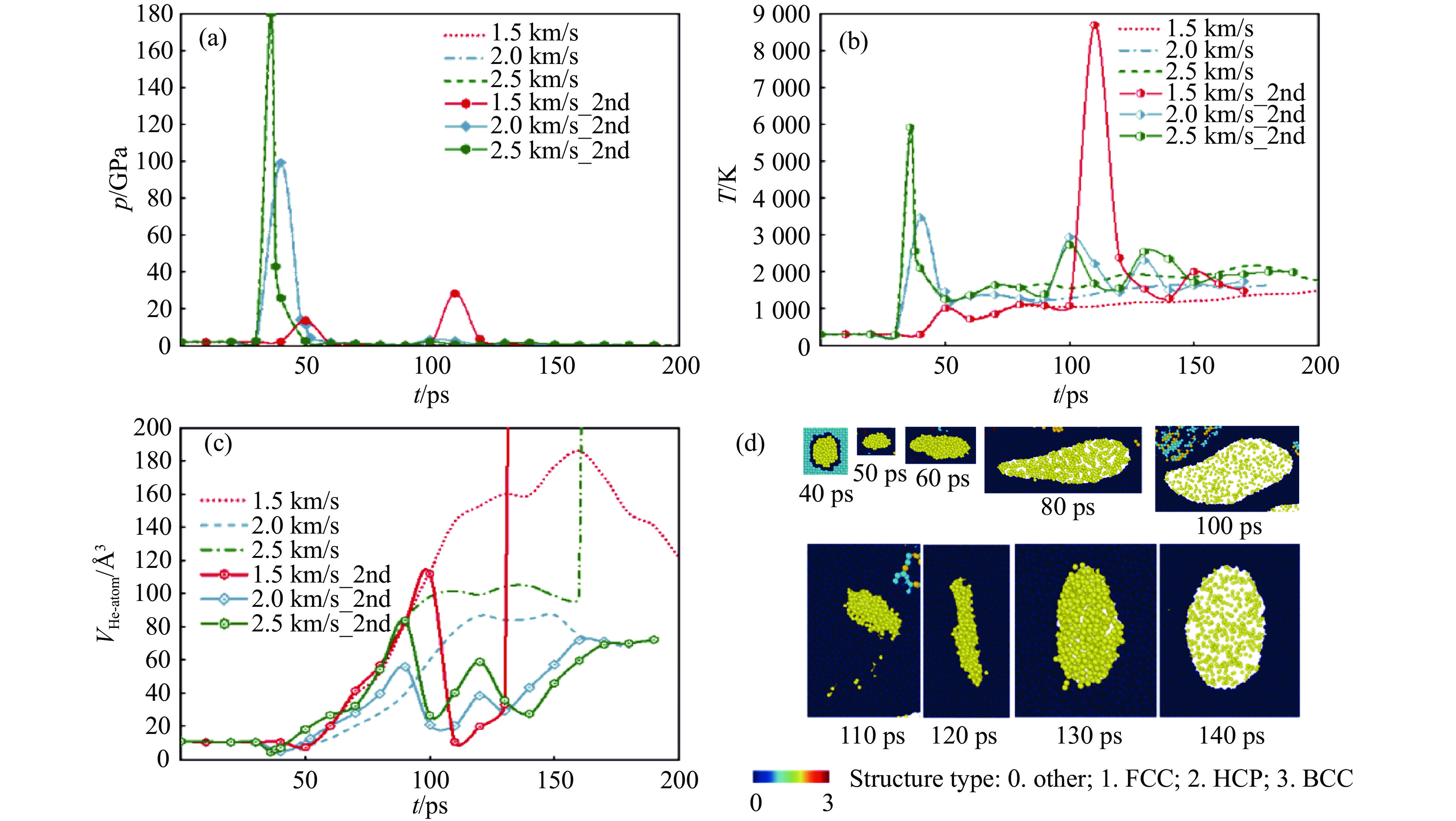
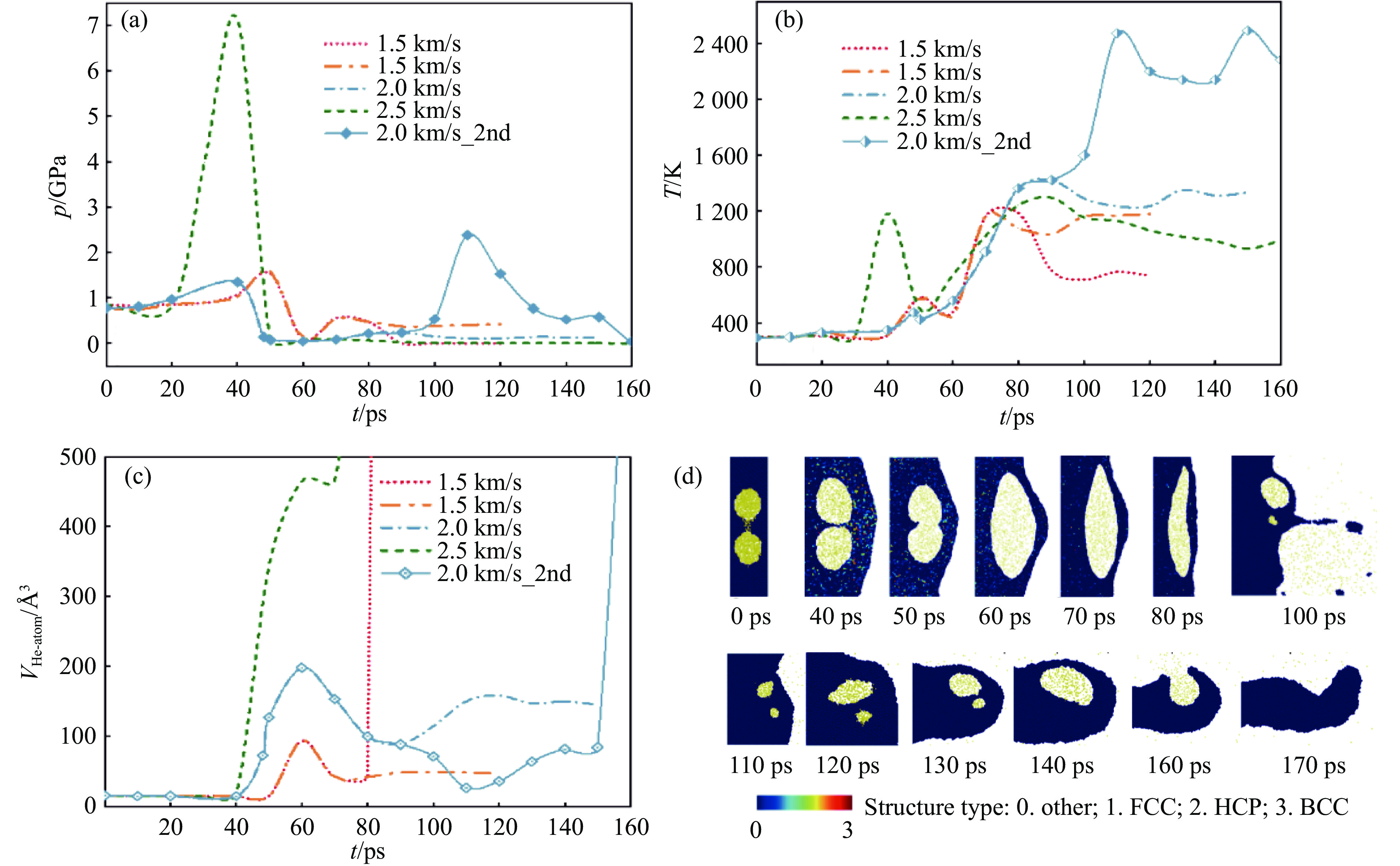
2026,
40(8): 080105.
doi: 10.11858/gywlxb.20251288
Abstract:
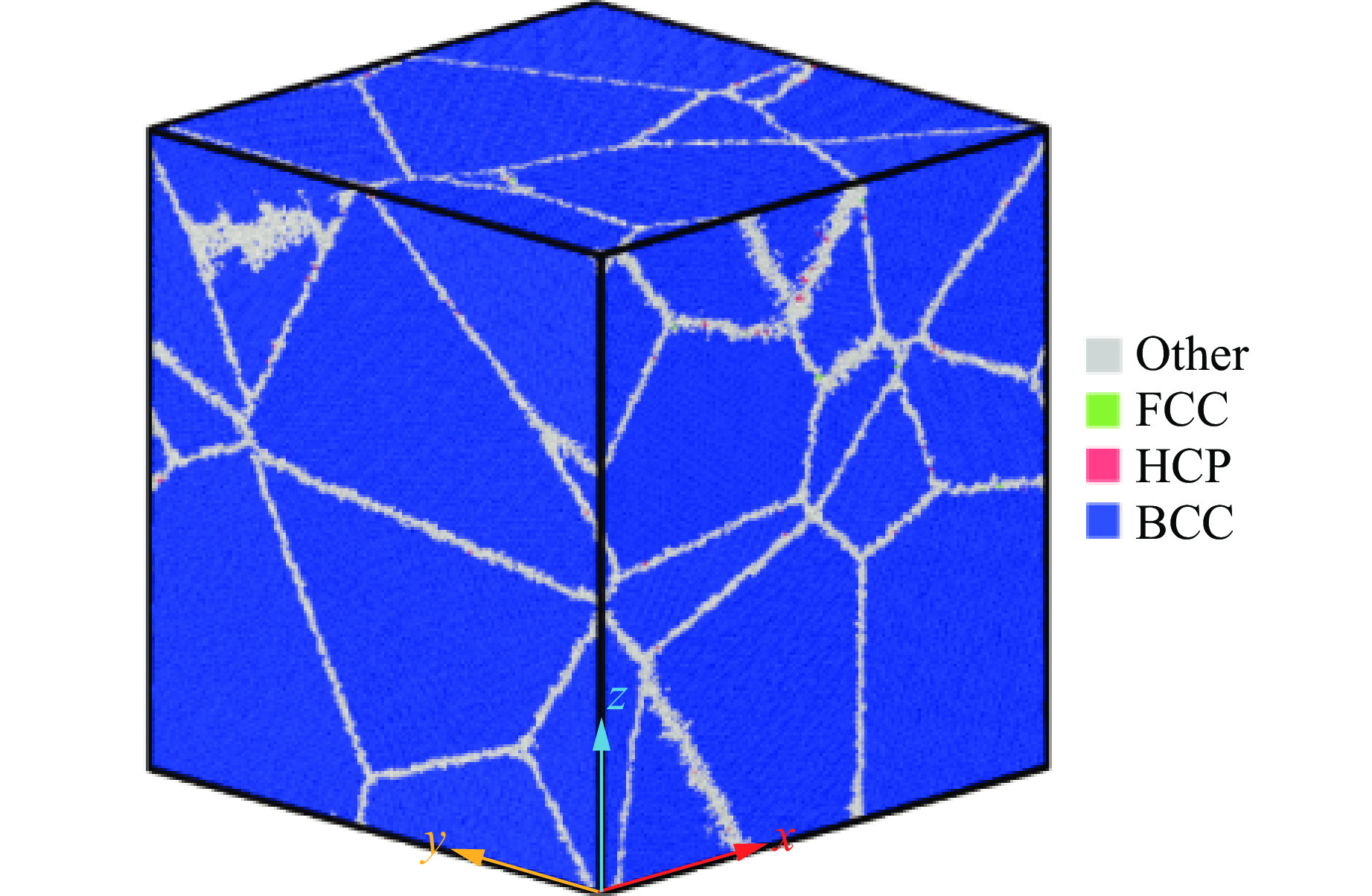
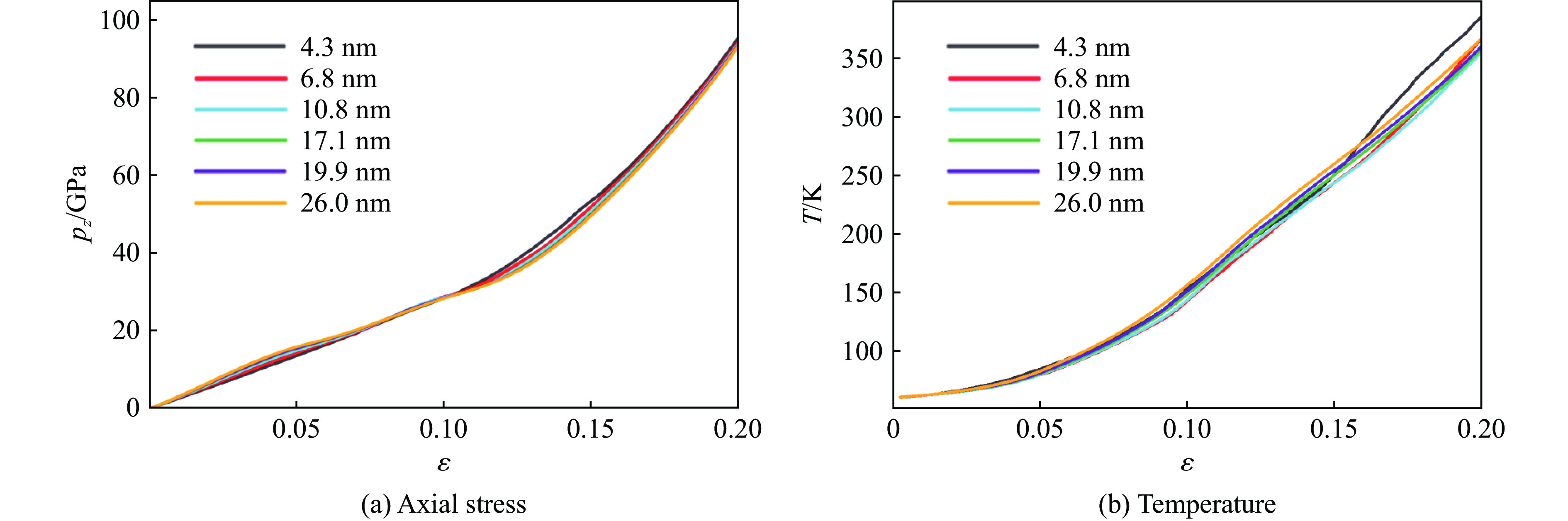
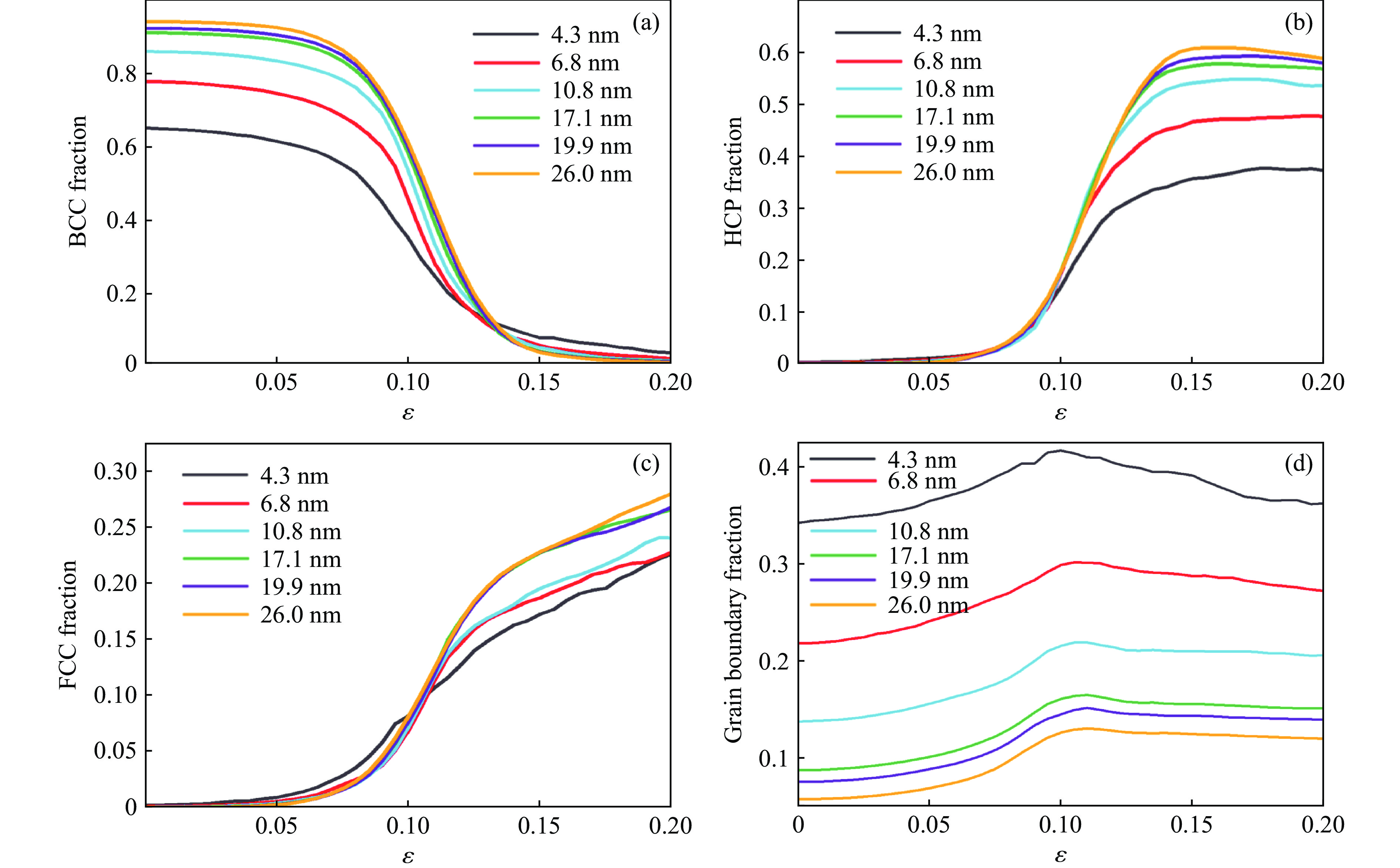
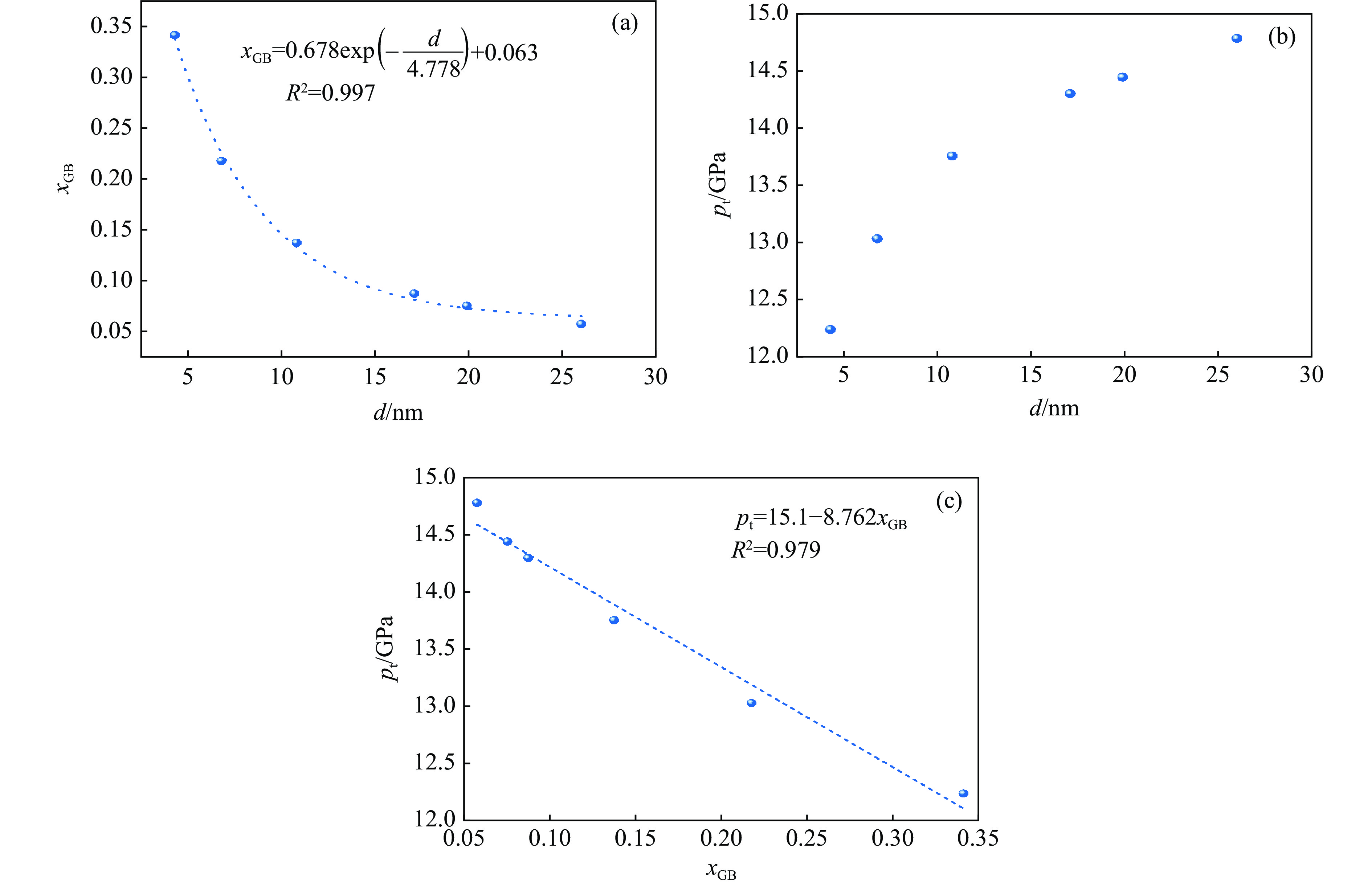
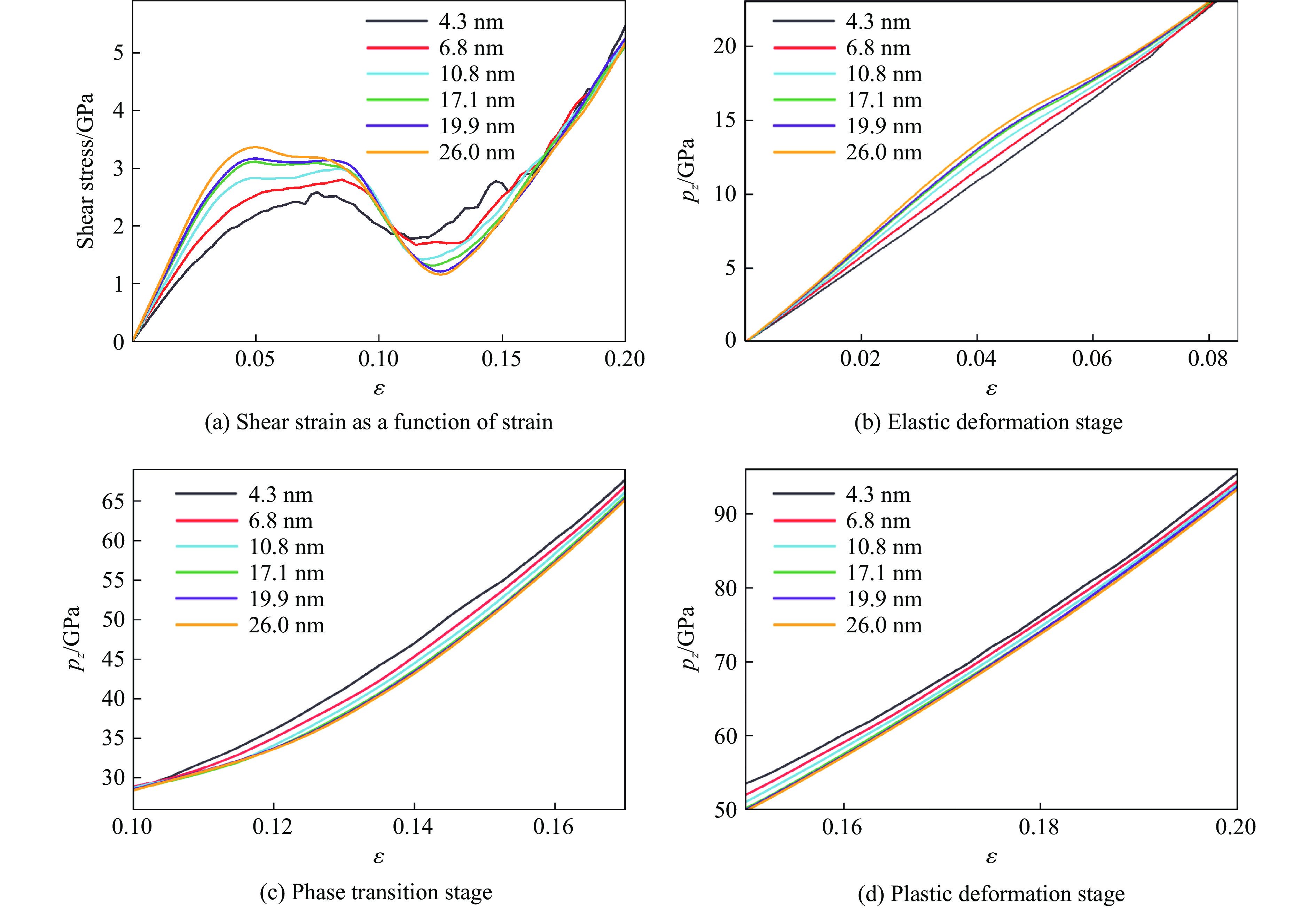
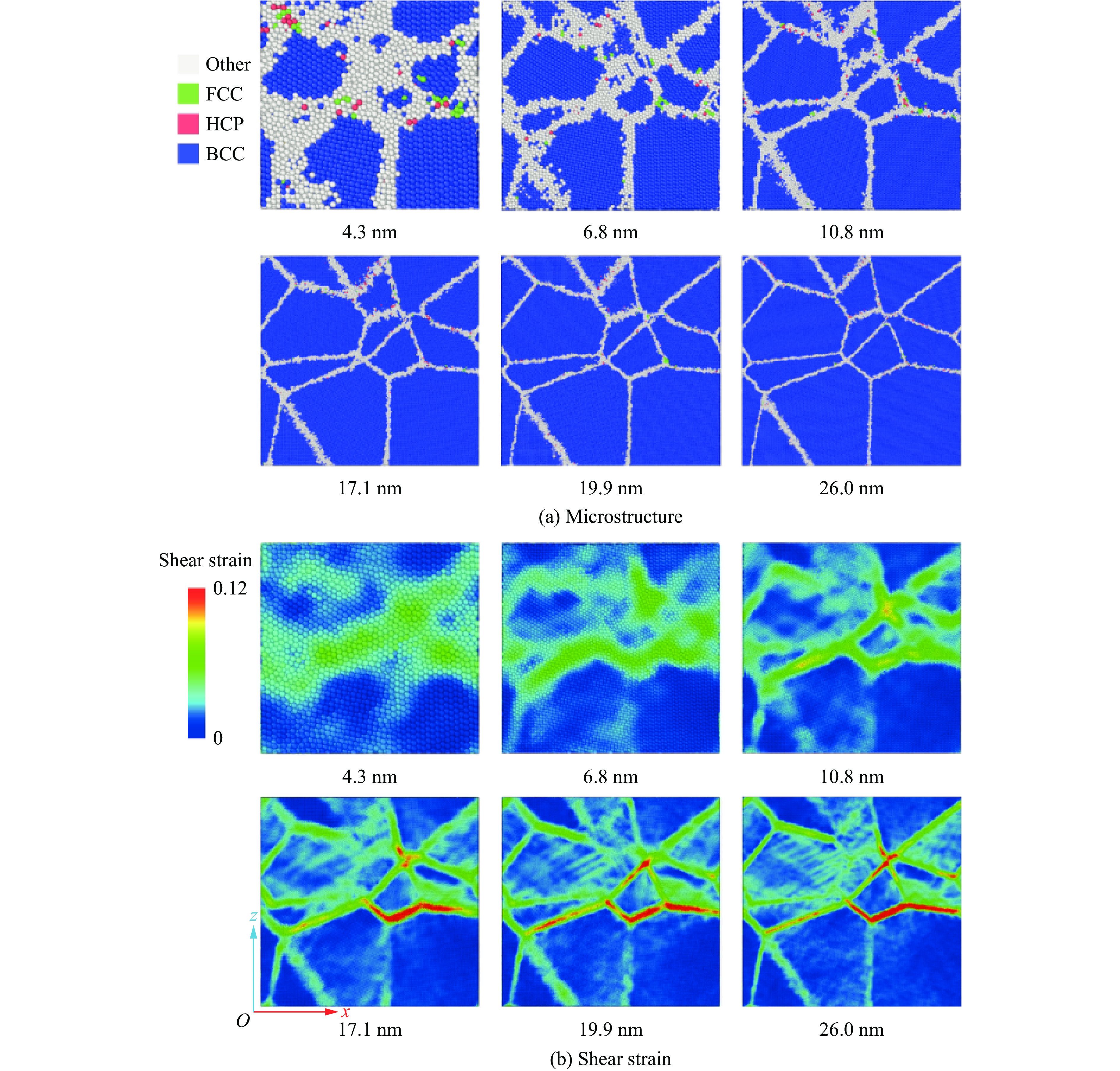

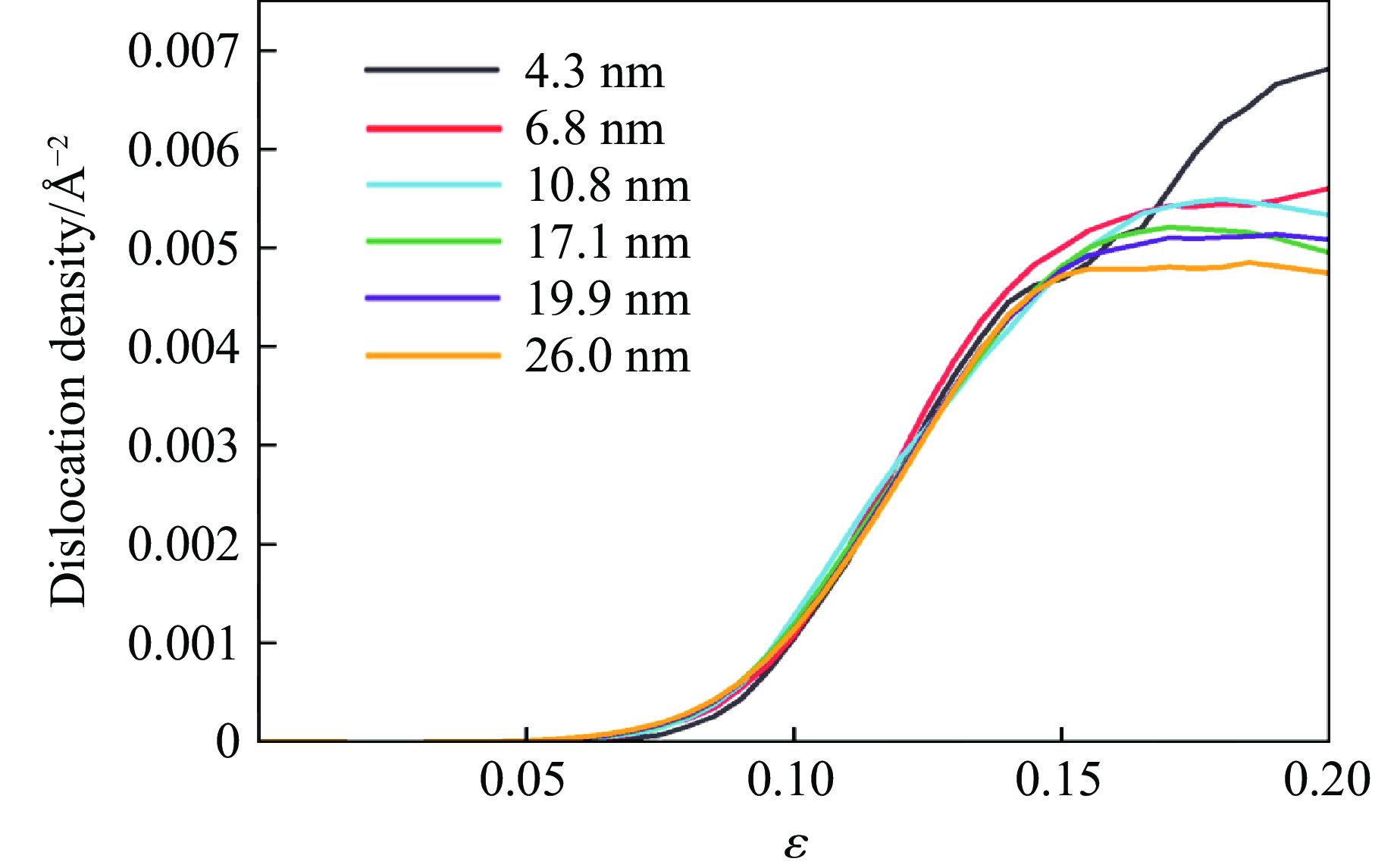

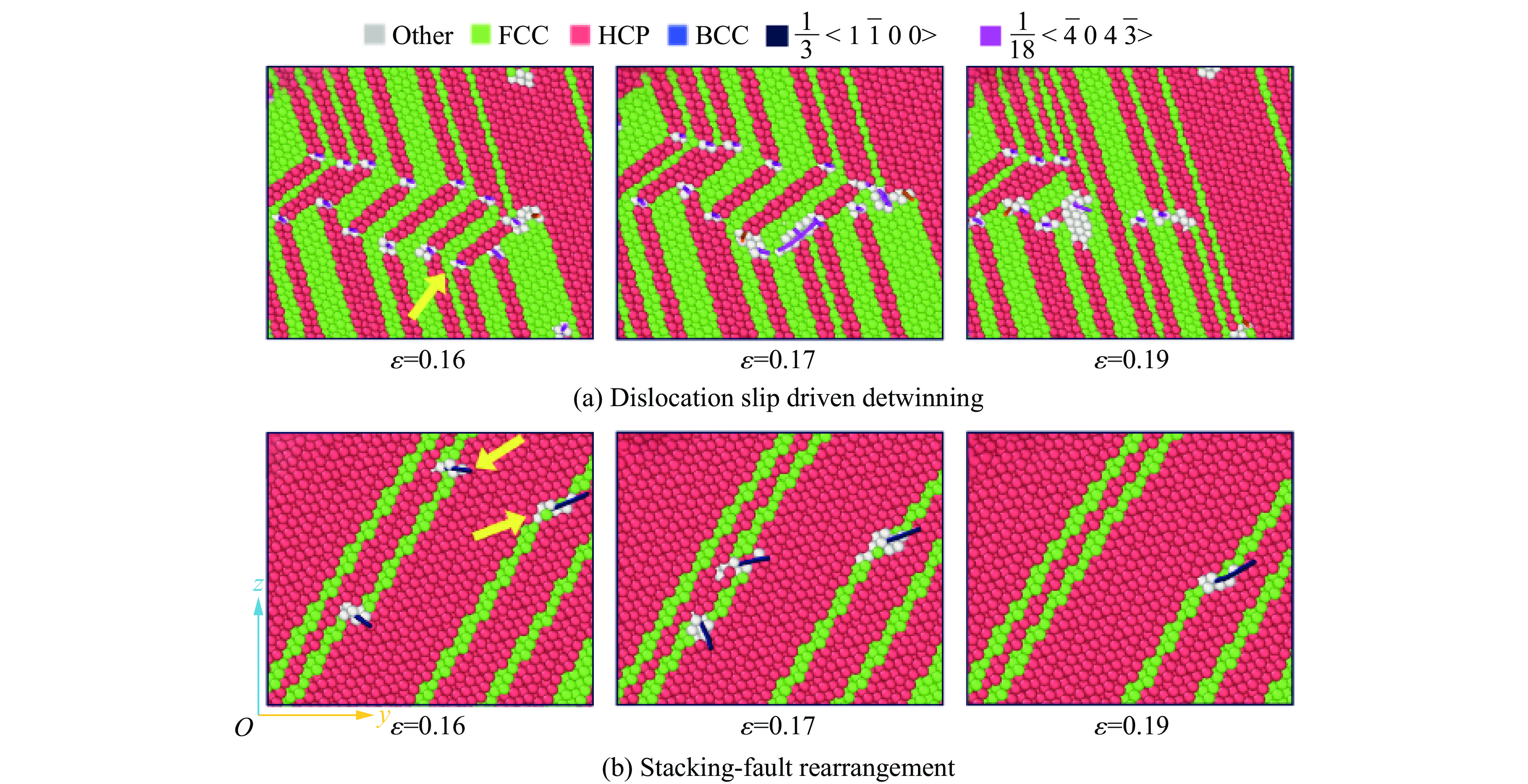
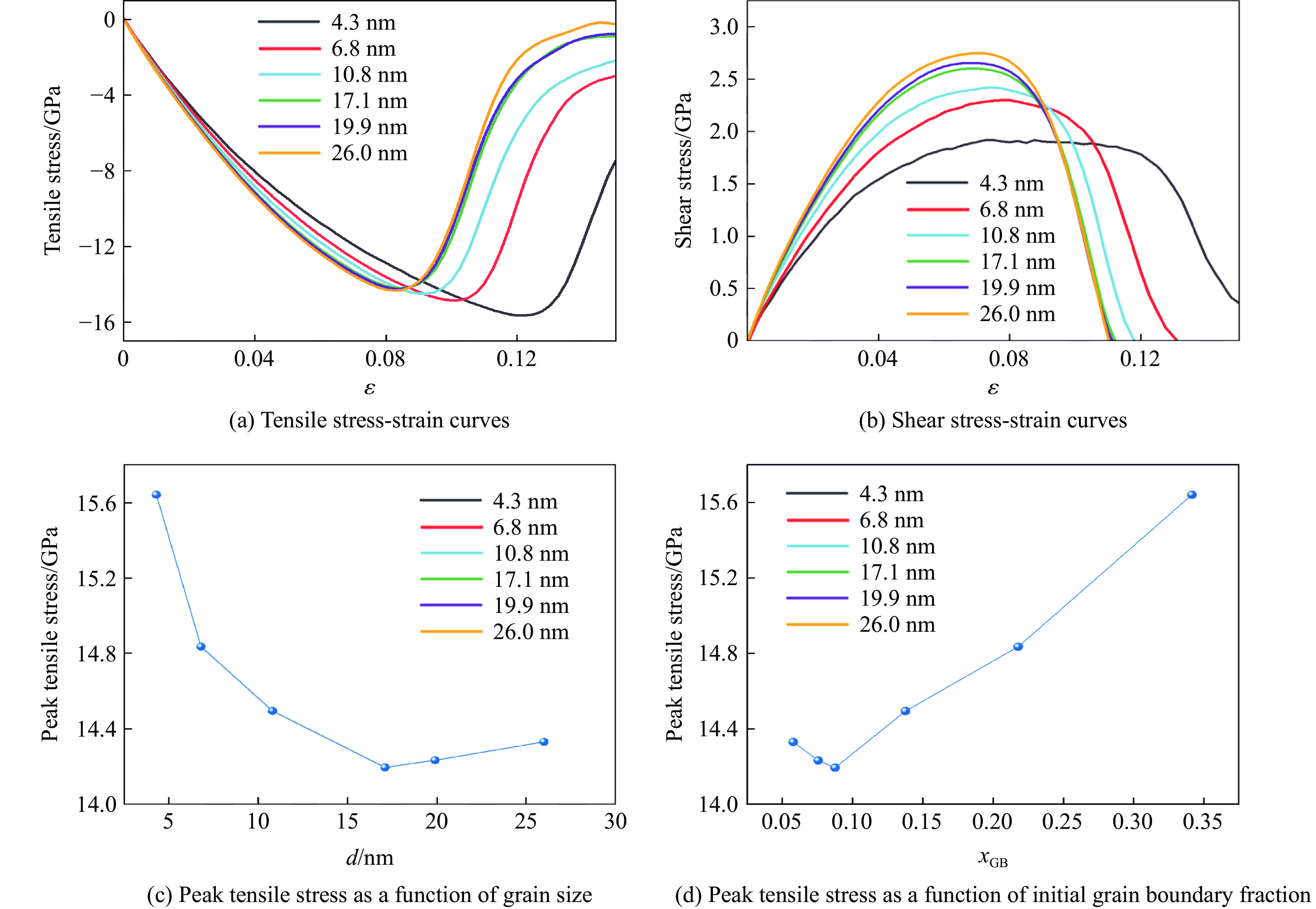

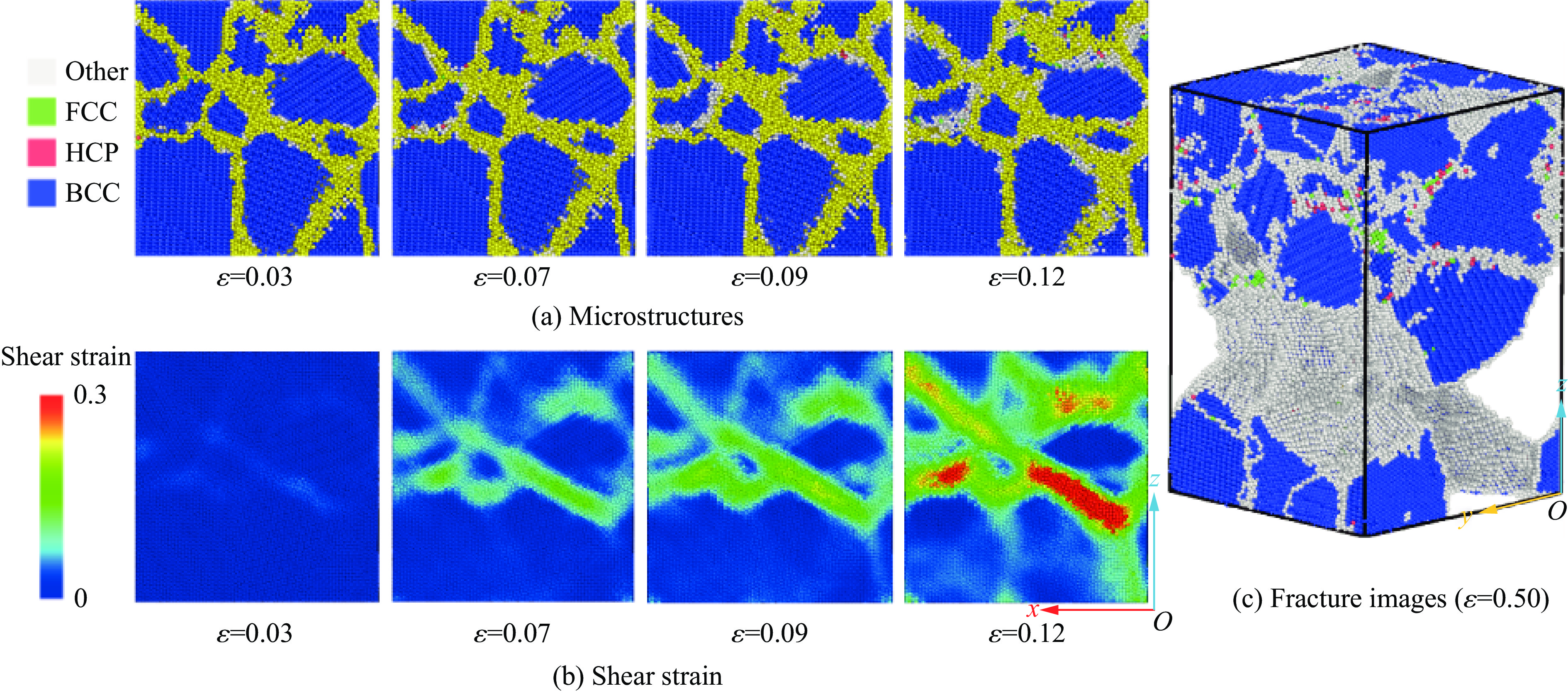
2026,
40(8): 080106.
doi: 10.11858/gywlxb.20251290
Abstract:
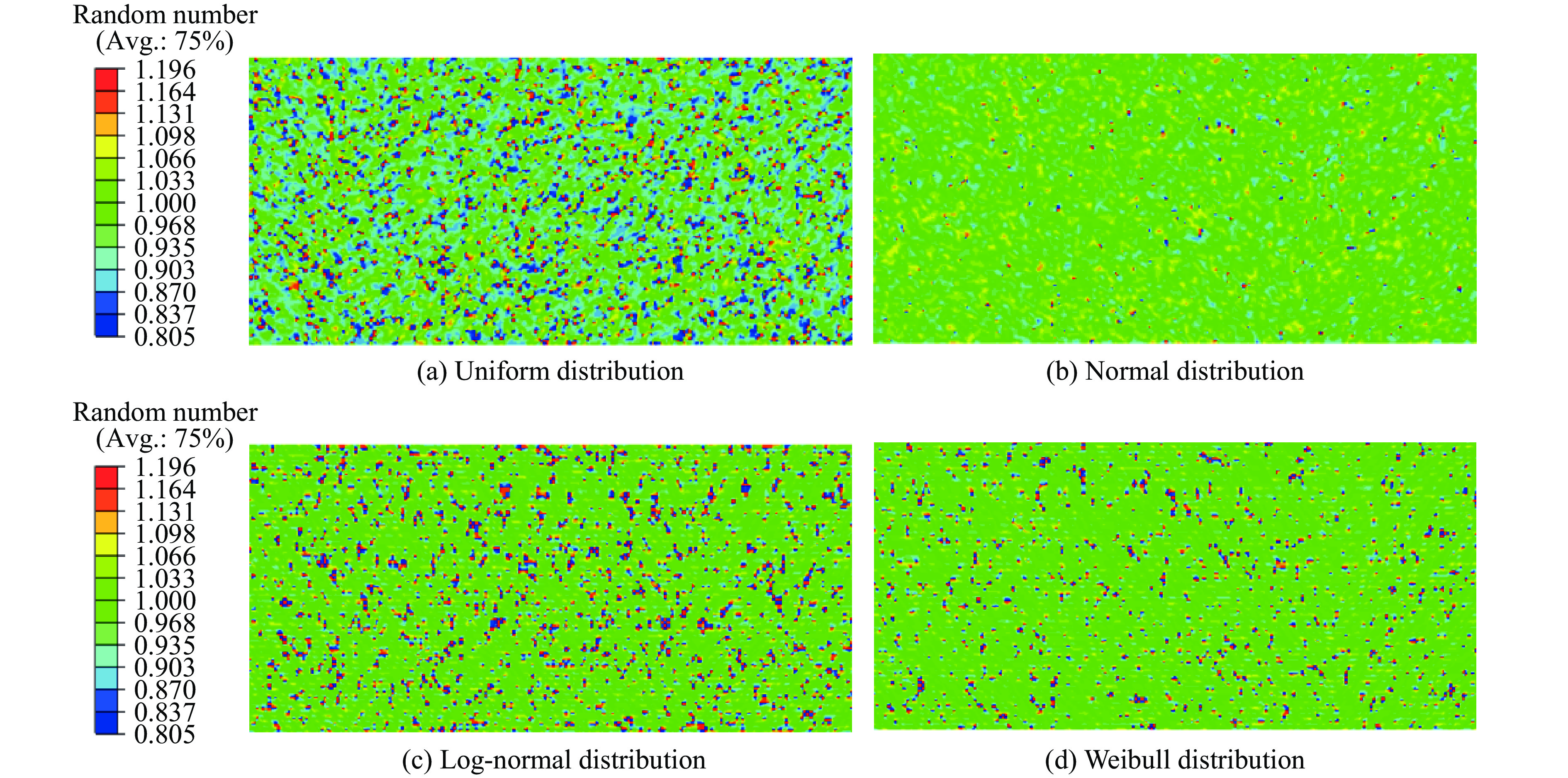
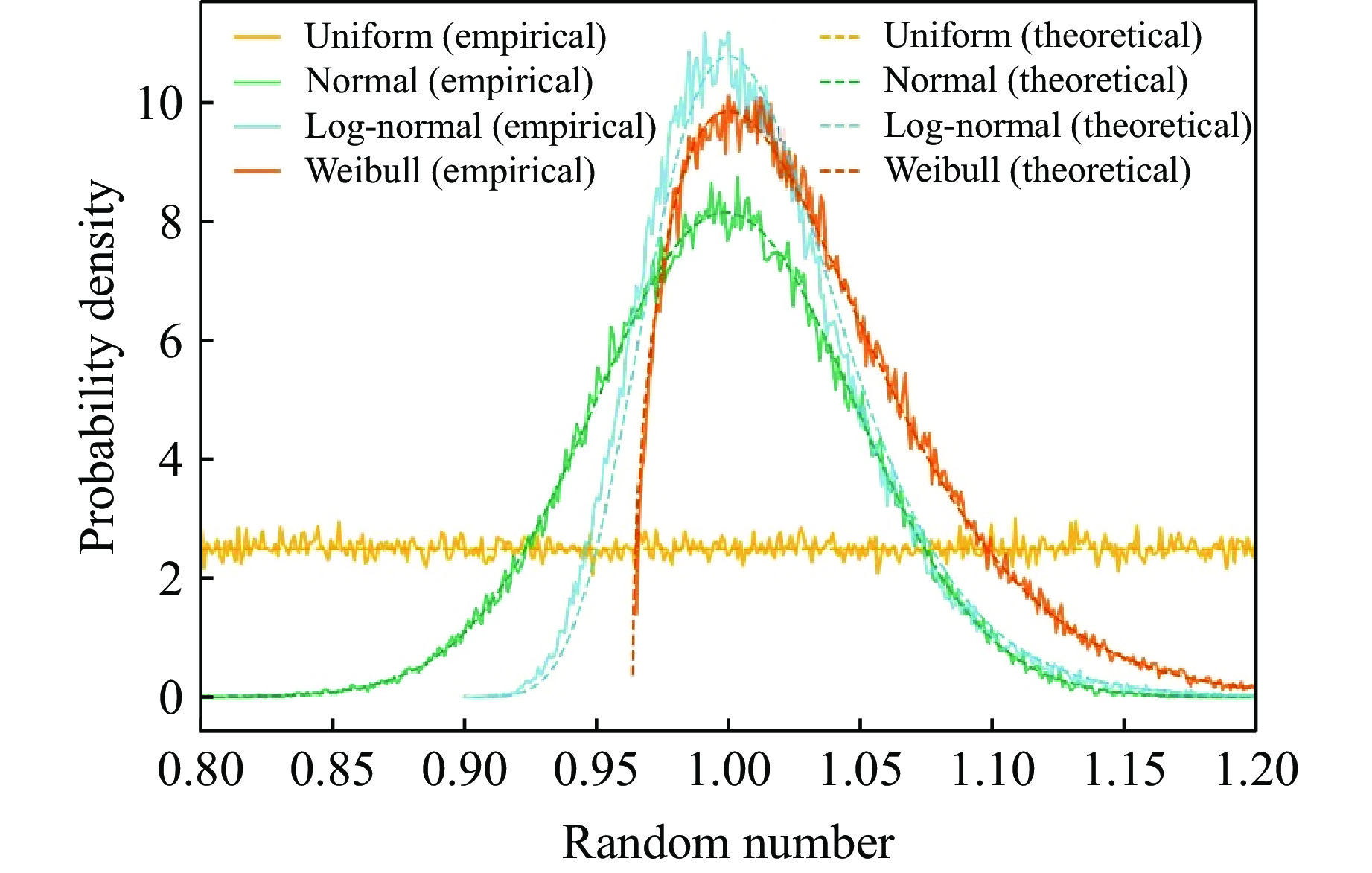
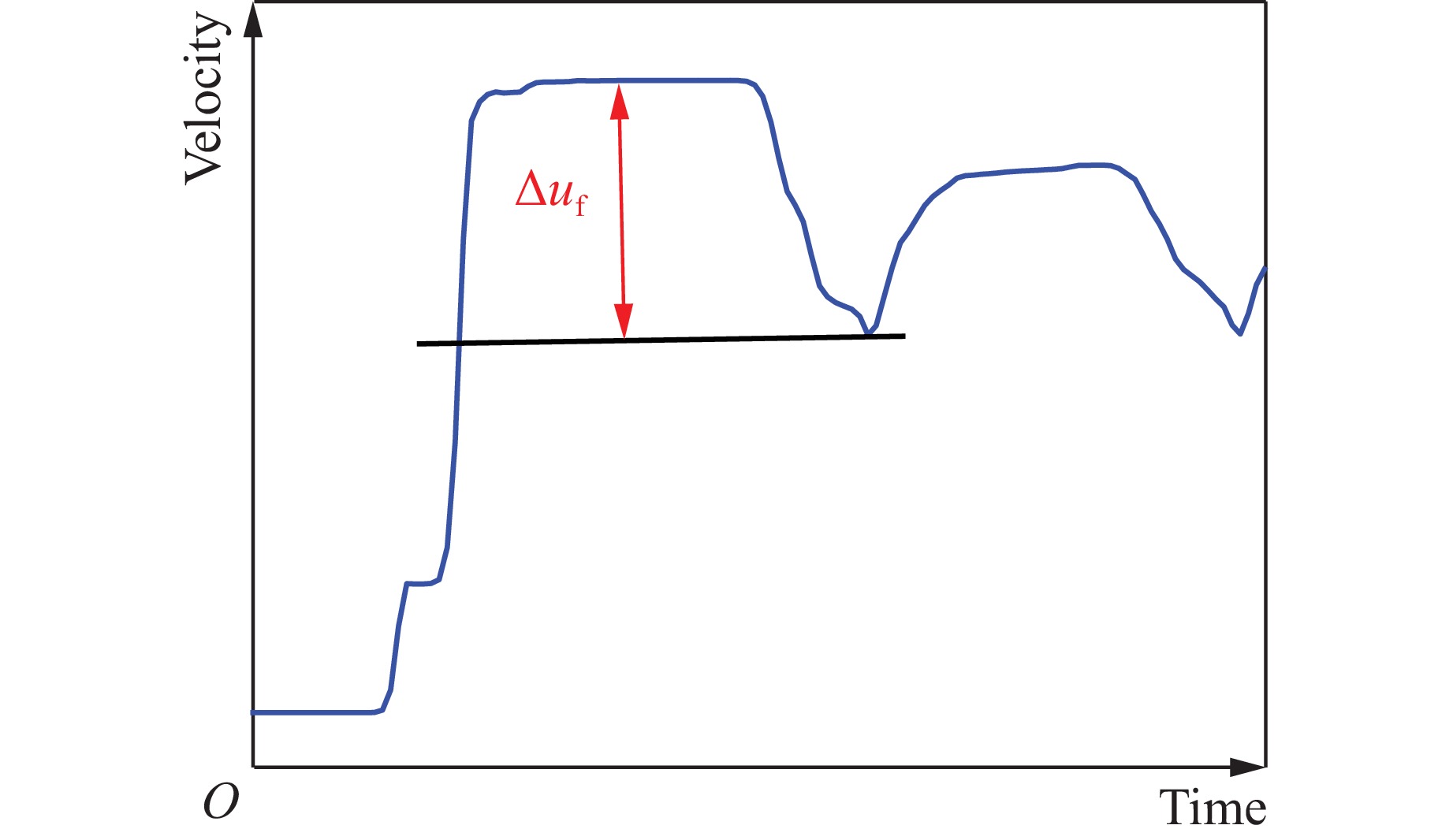
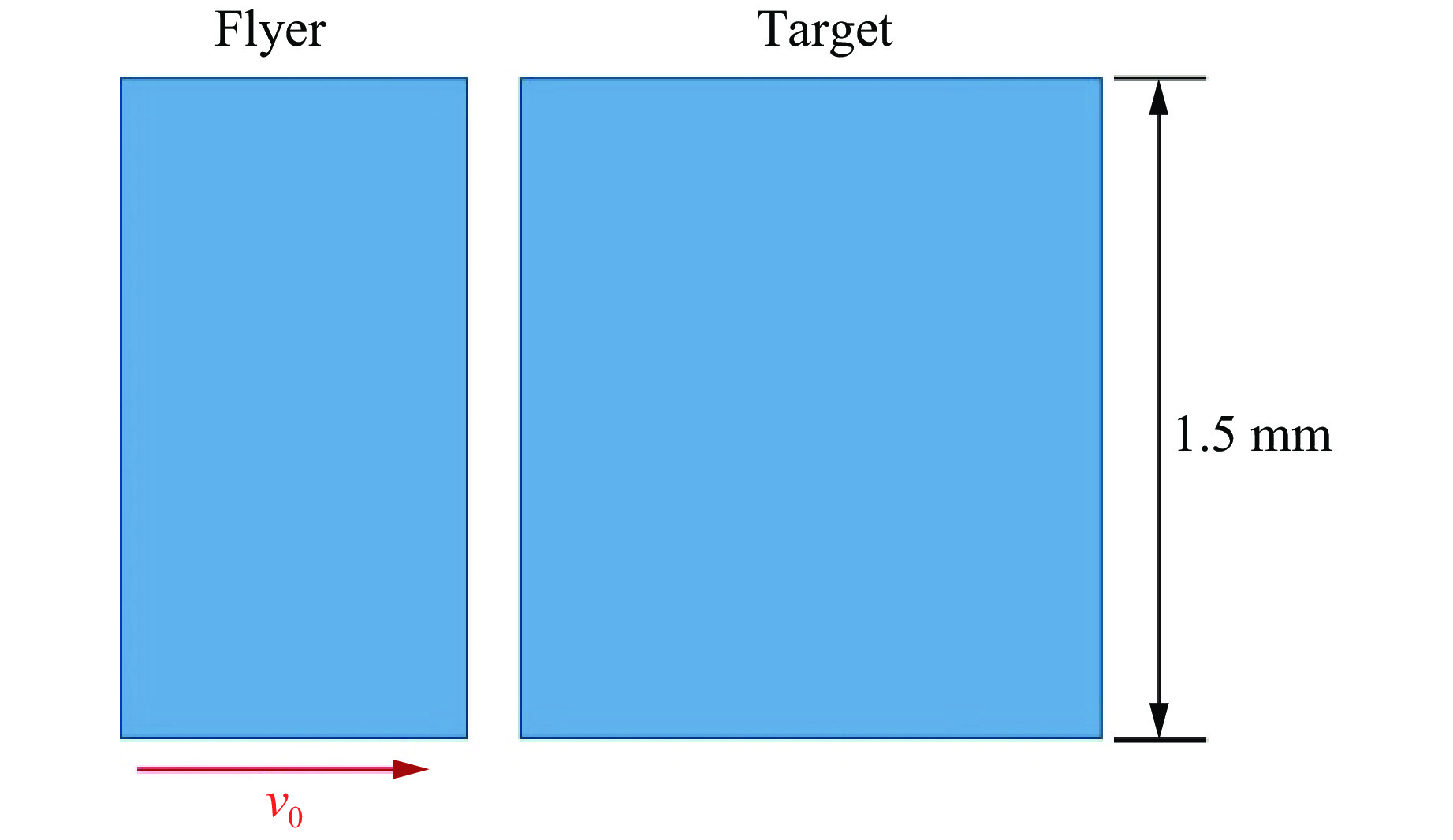
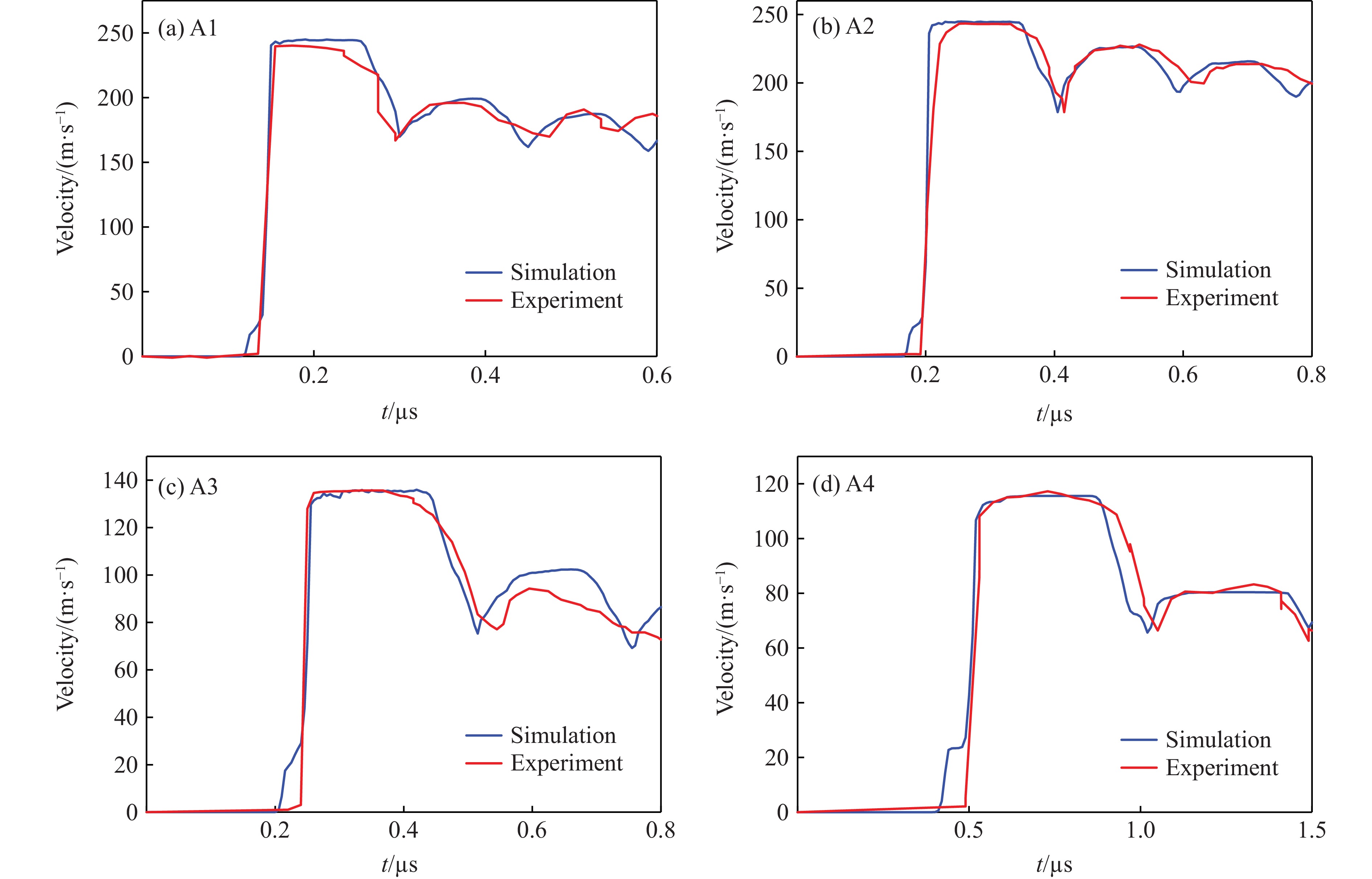
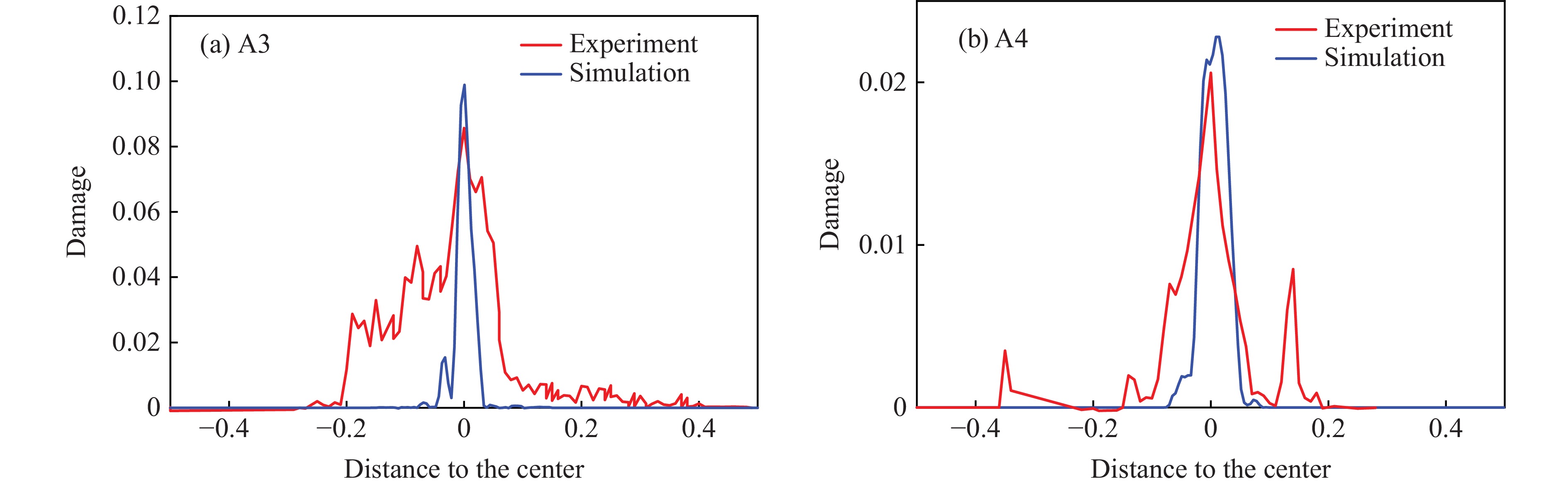
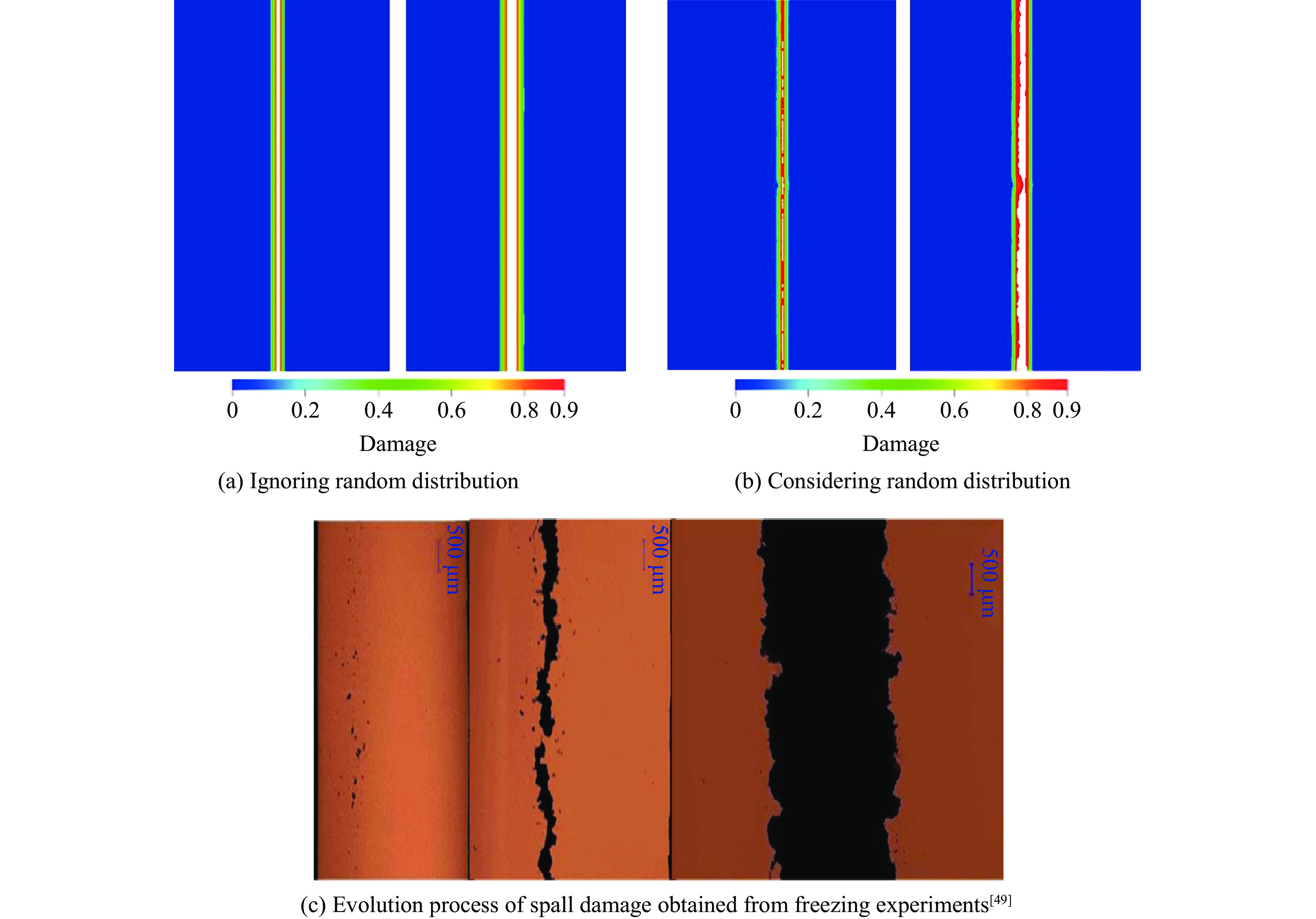
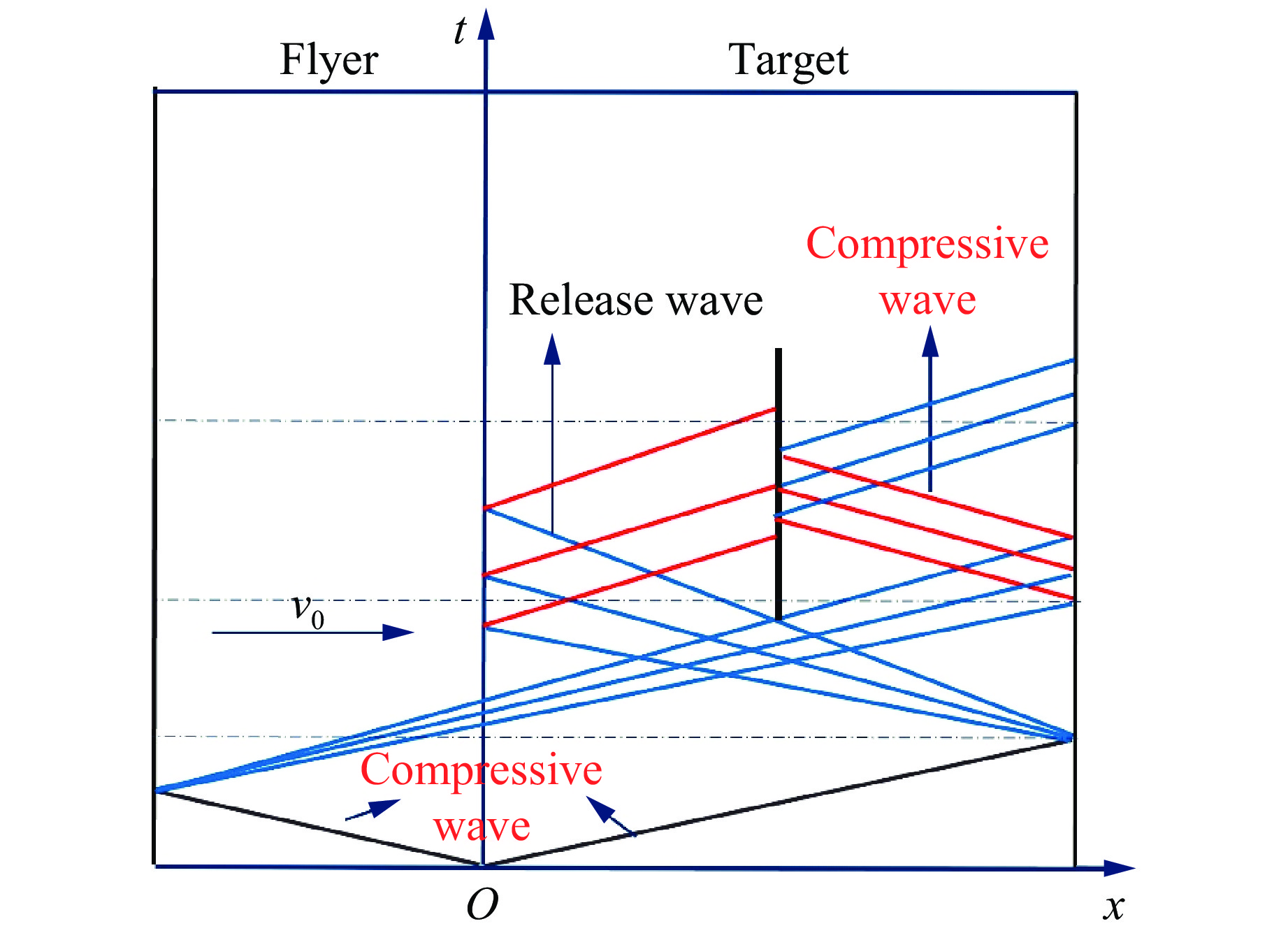
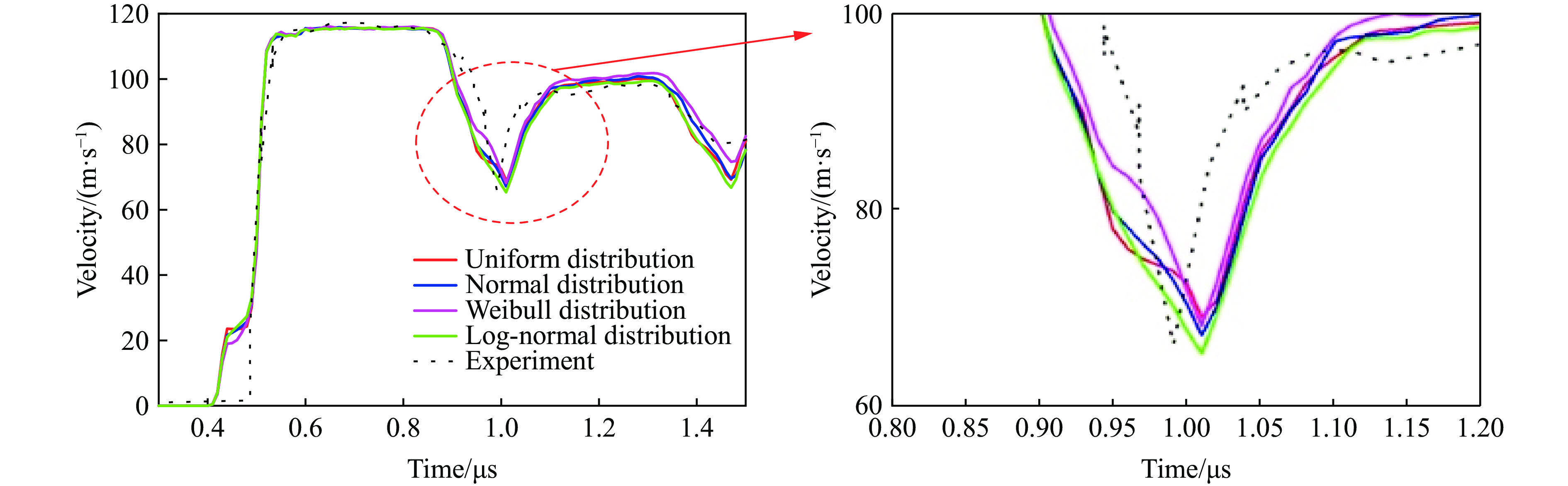
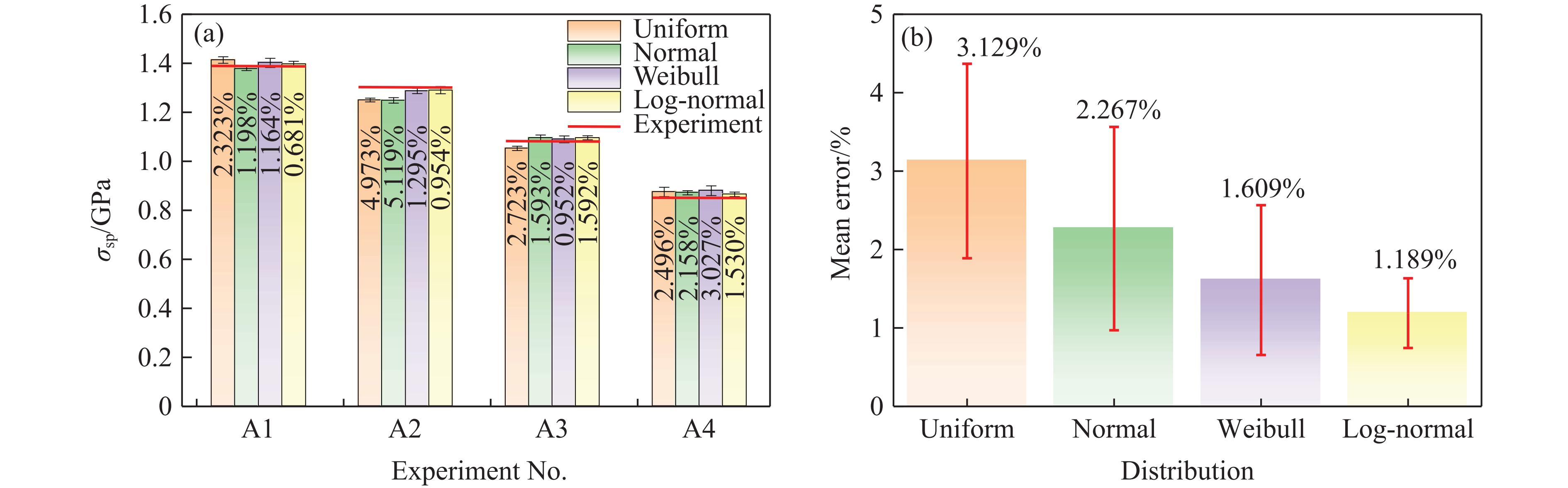
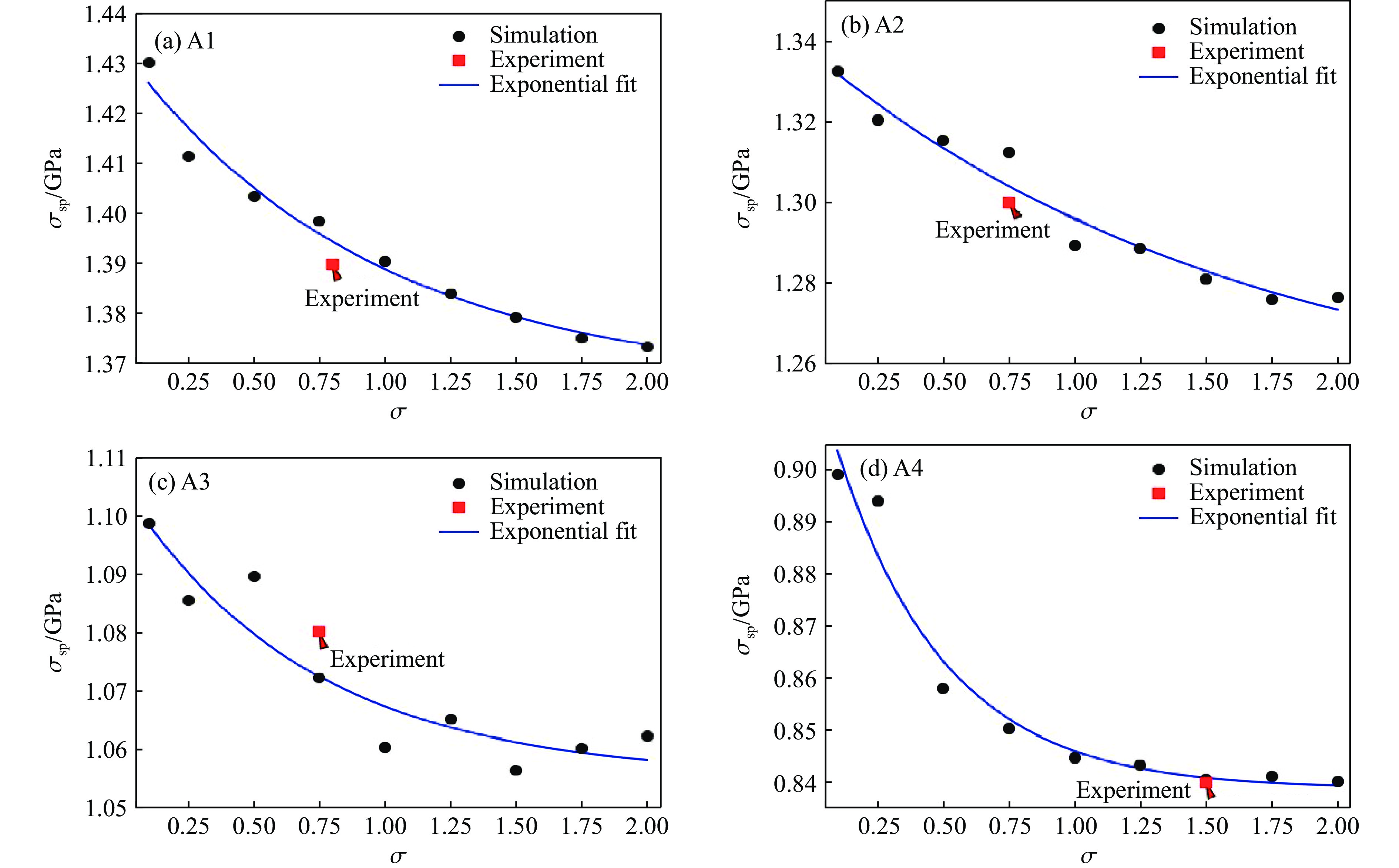
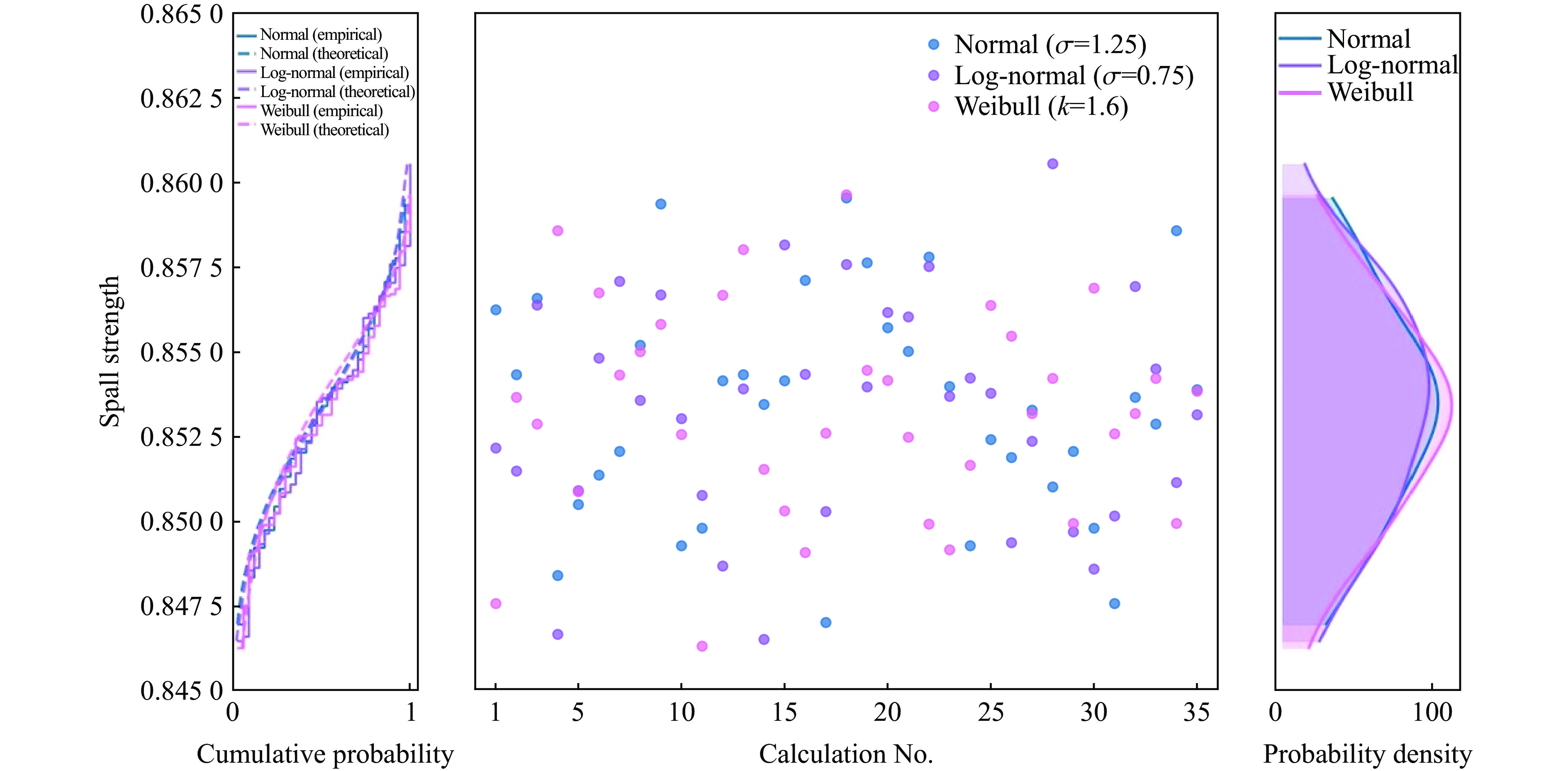
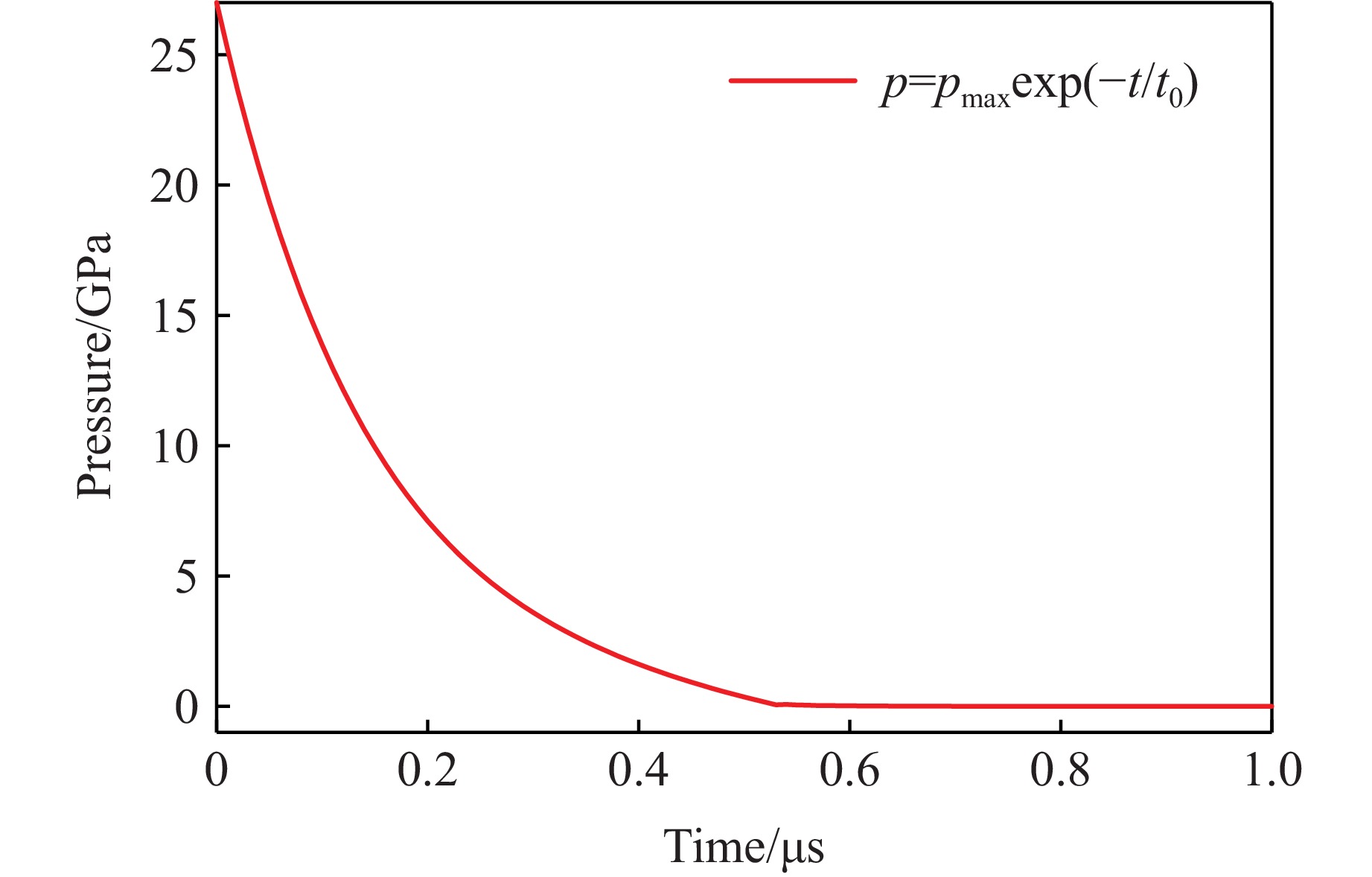
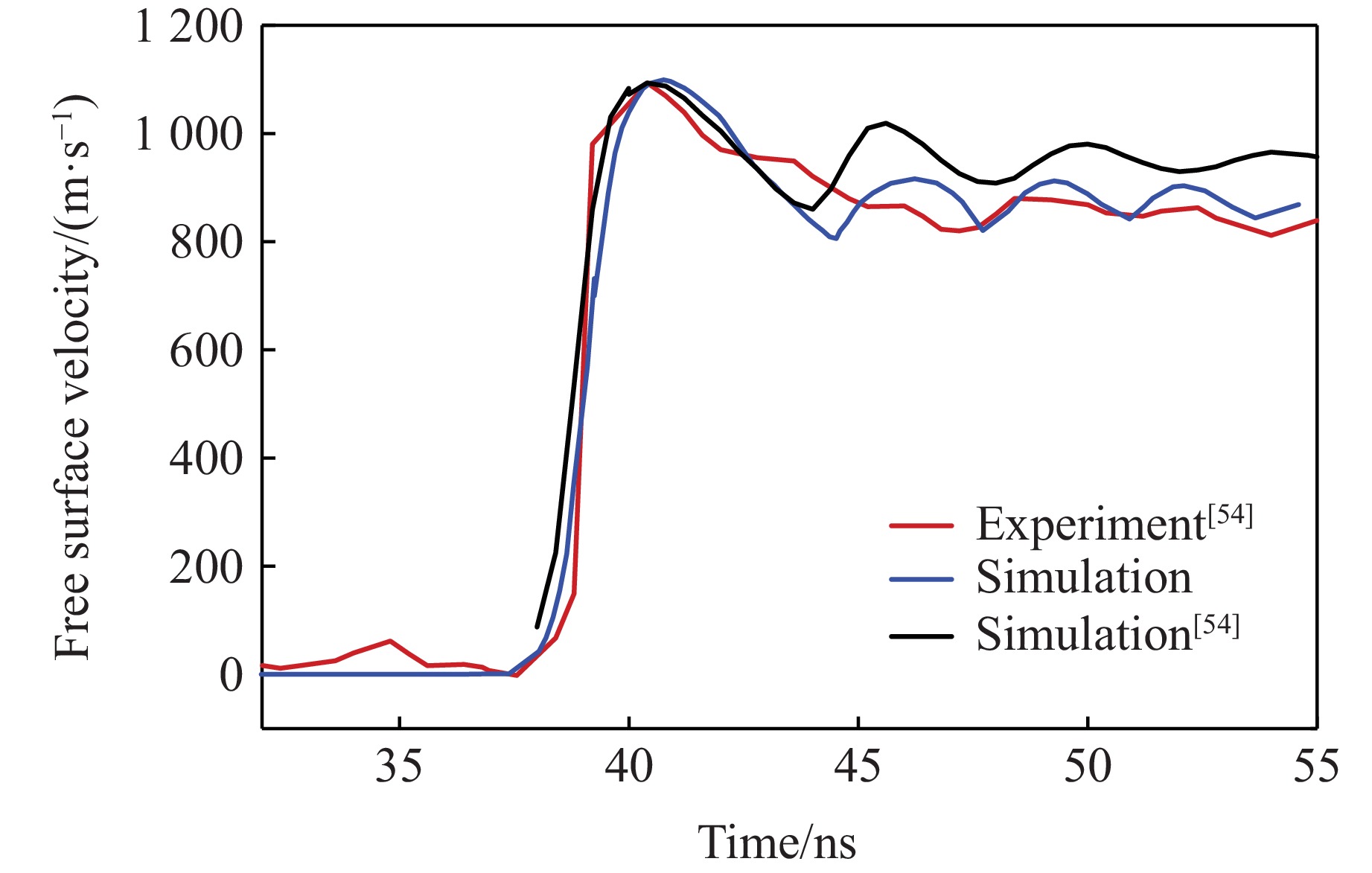
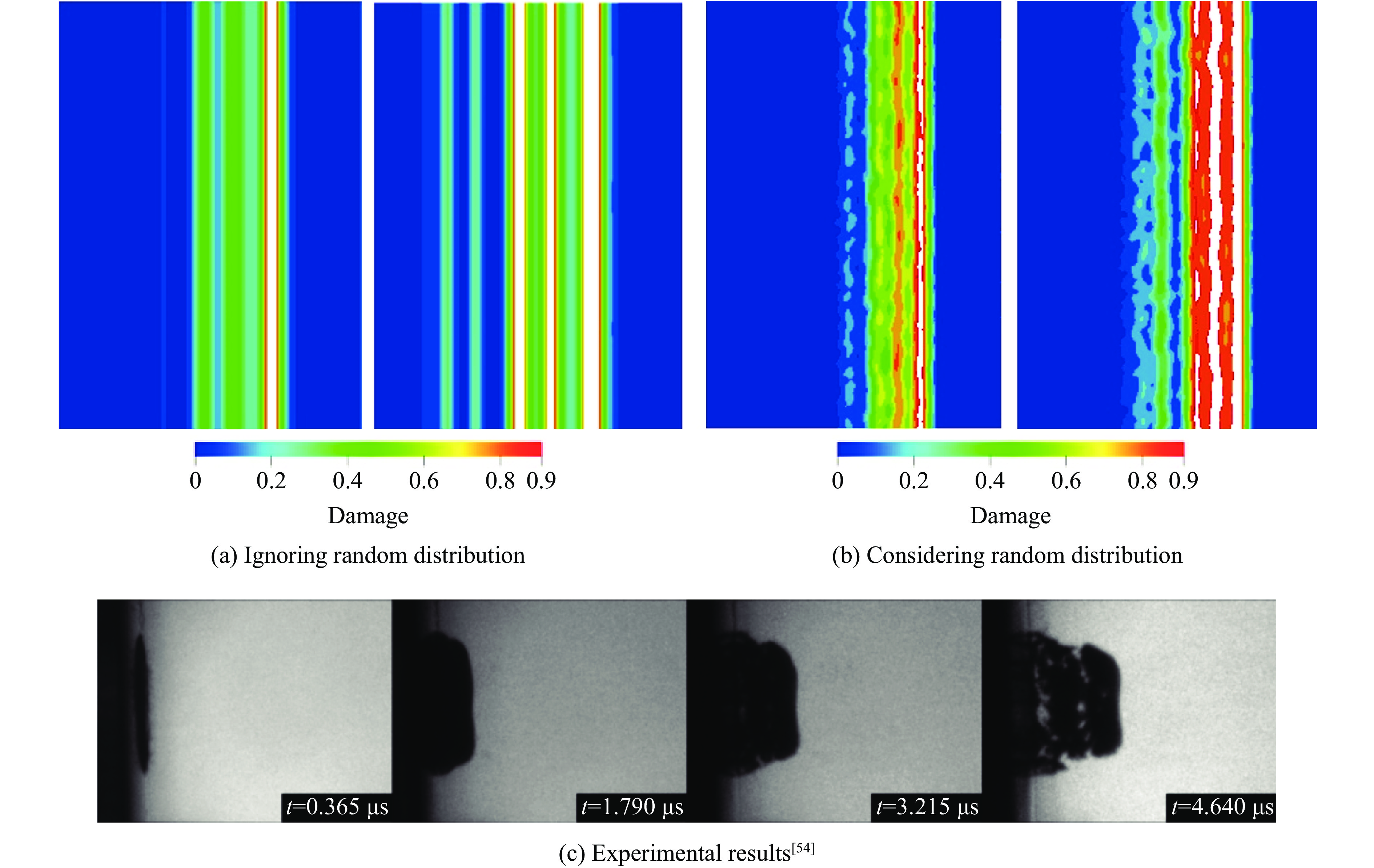
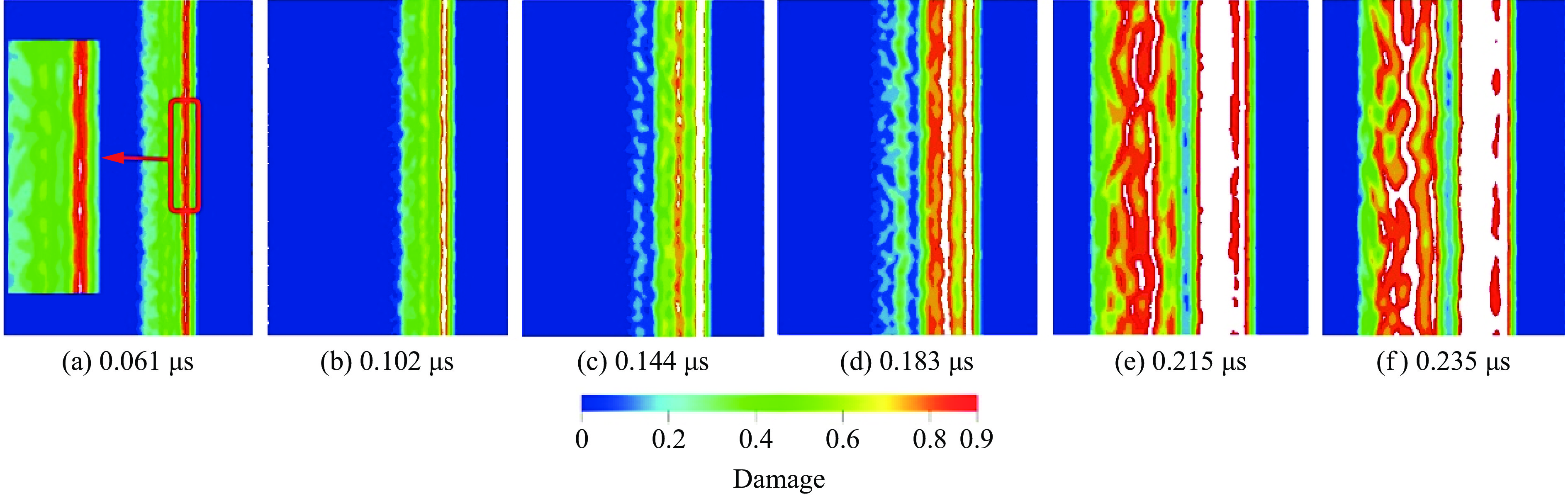
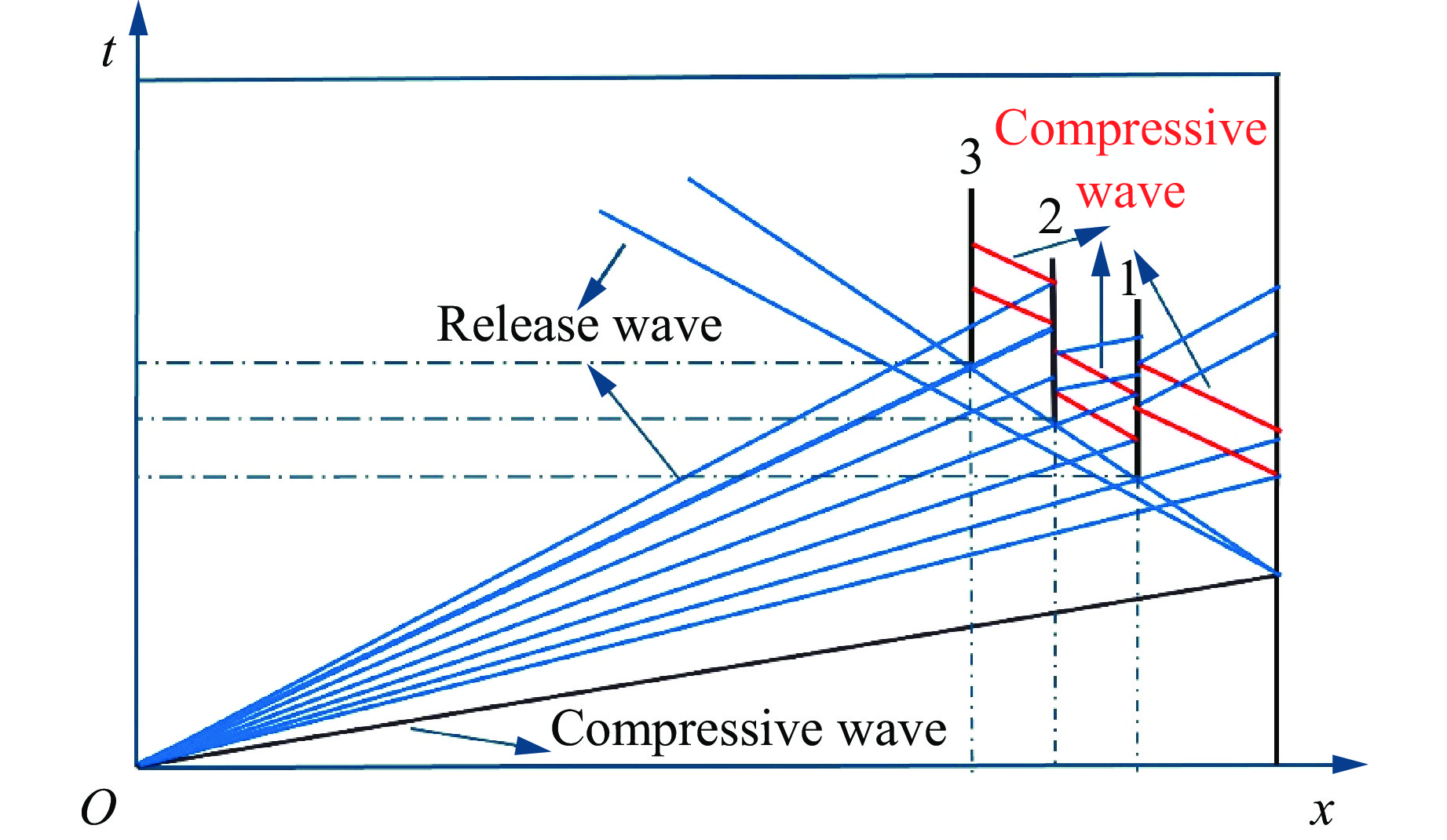
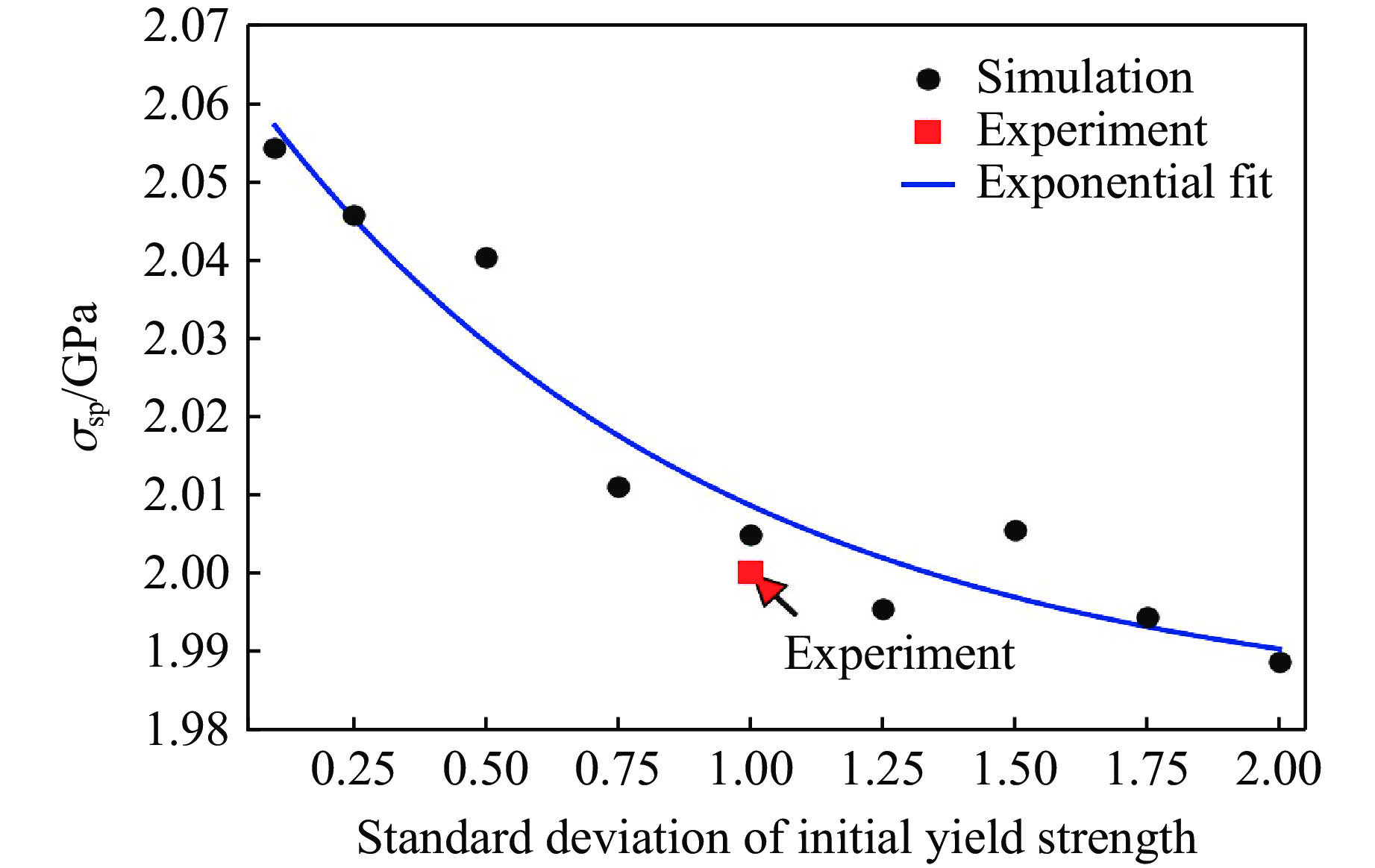

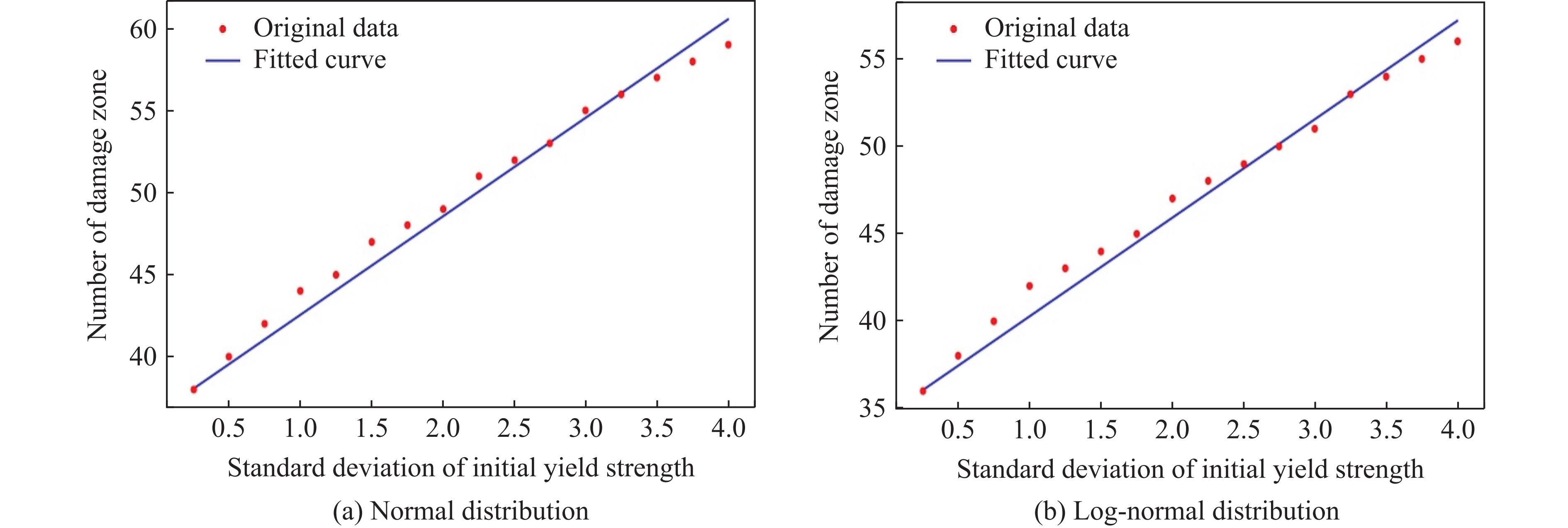
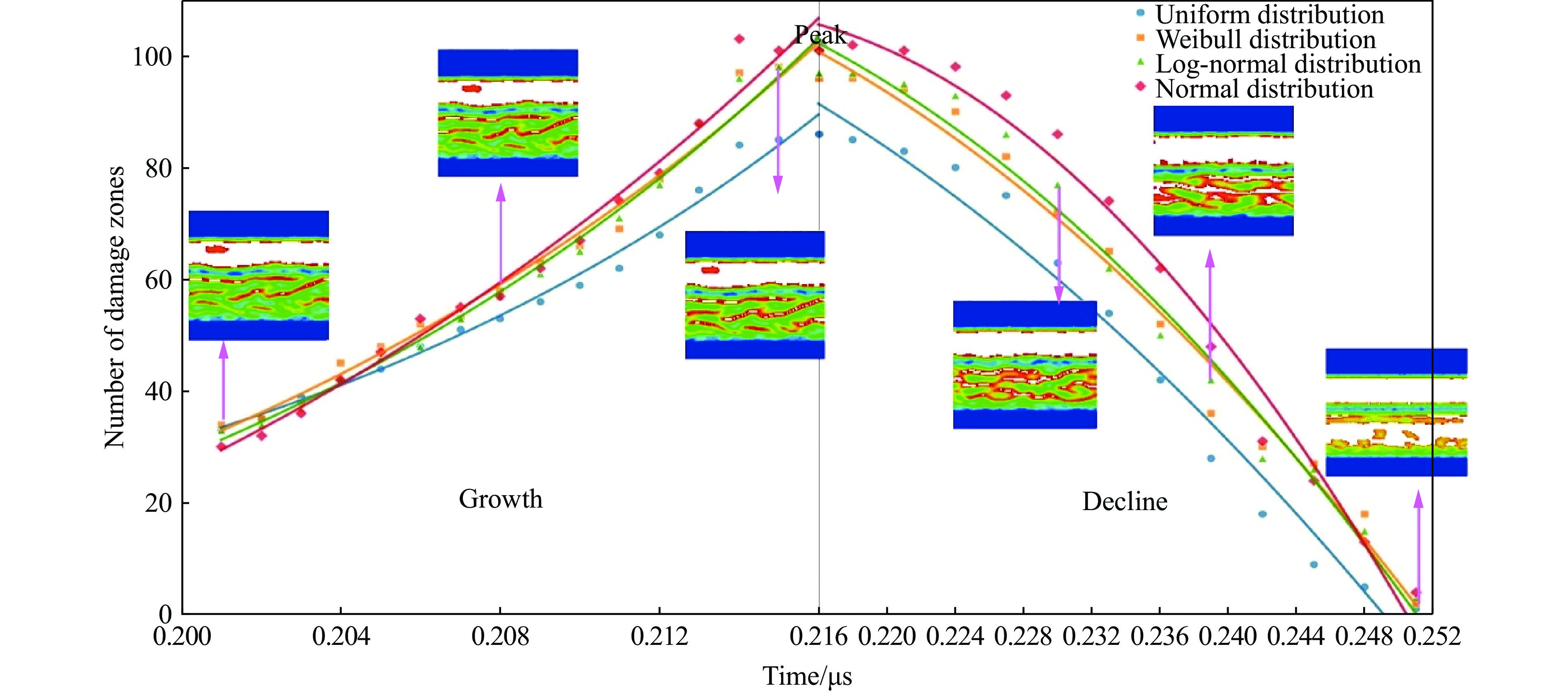
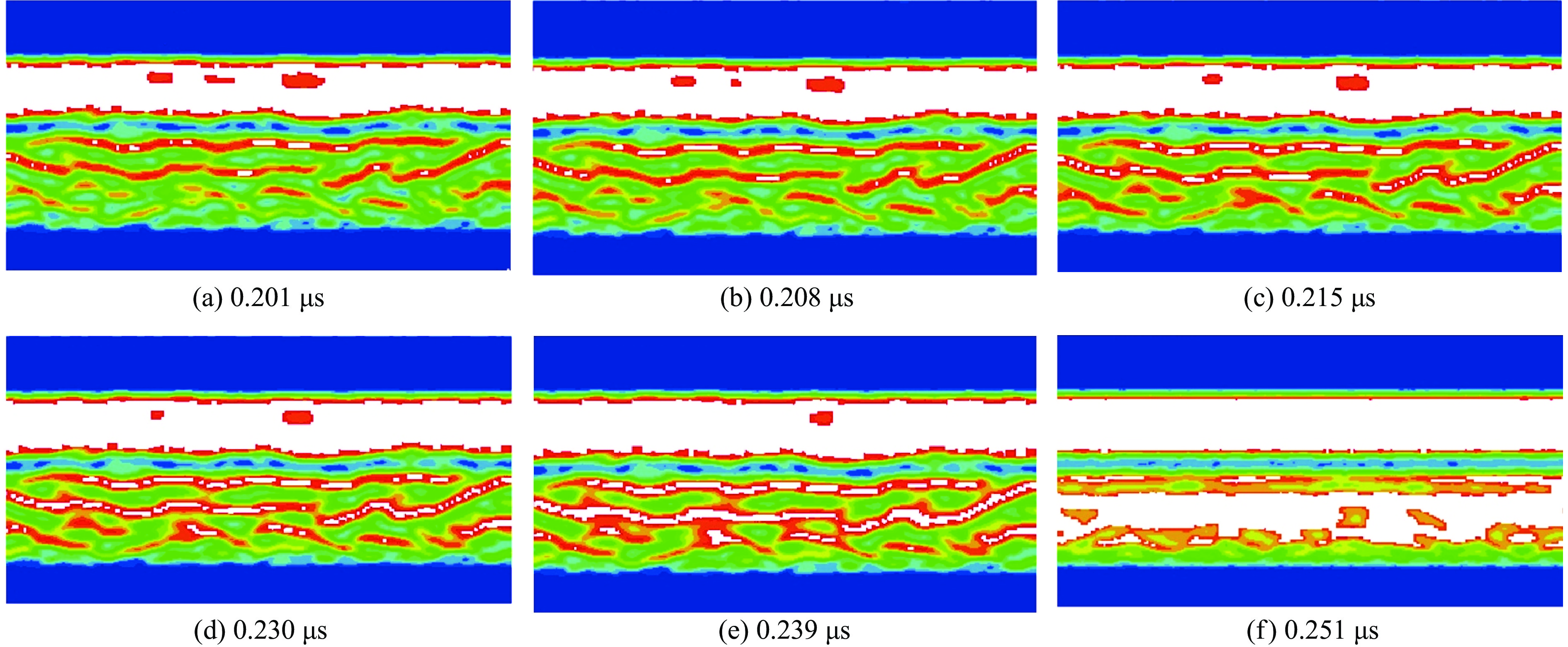
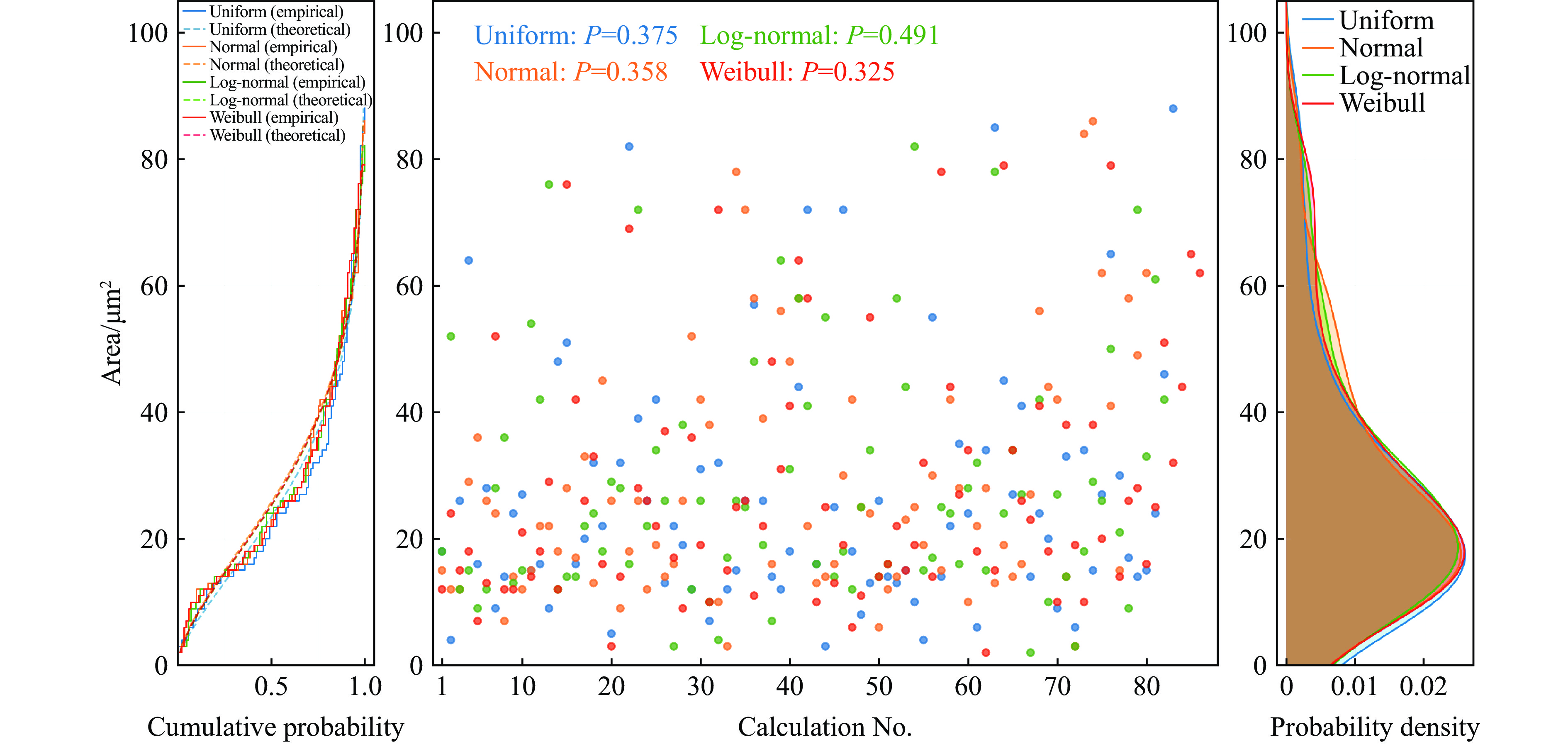
2026,
40(8): 080107.
doi: 10.11858/gywlxb.20251057
Abstract:
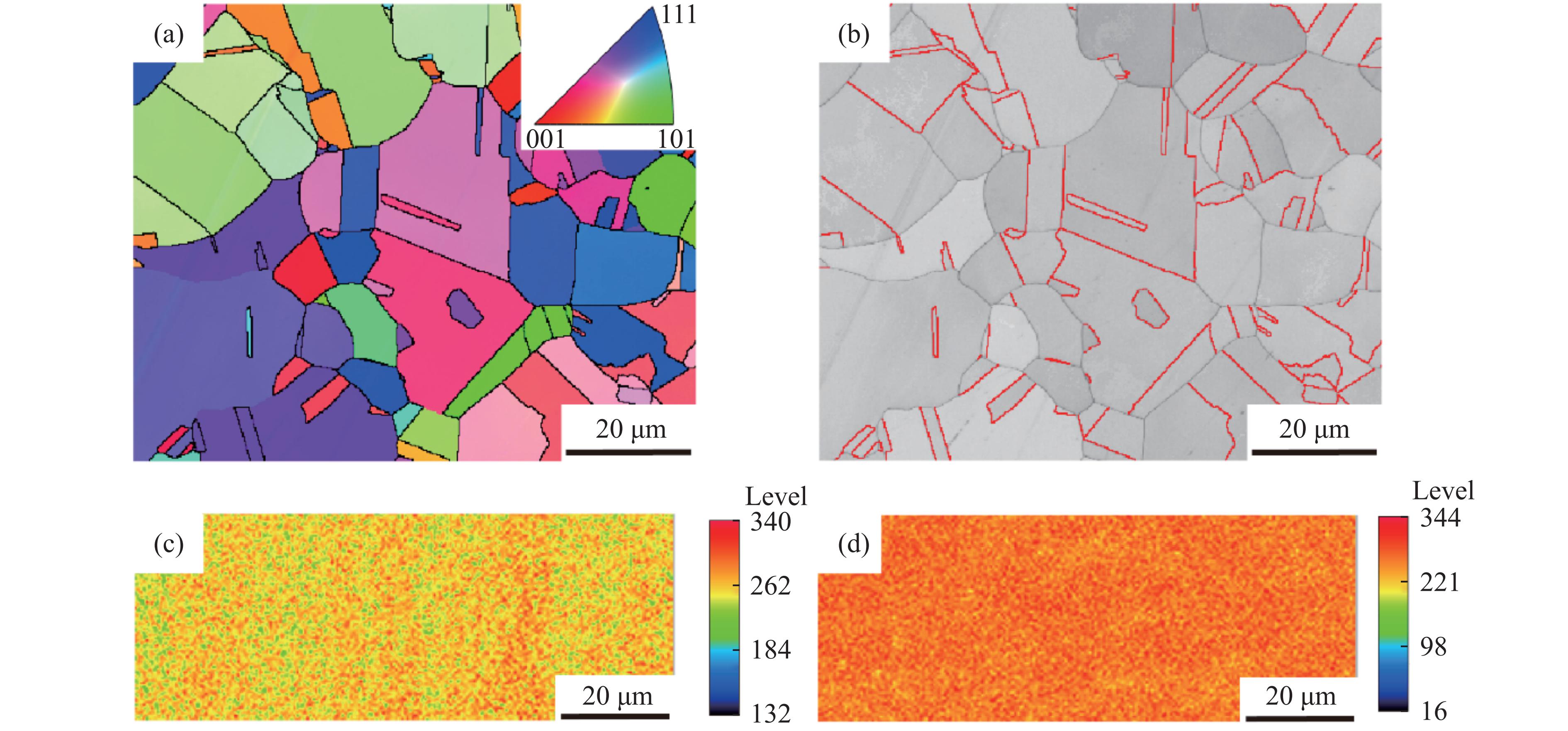
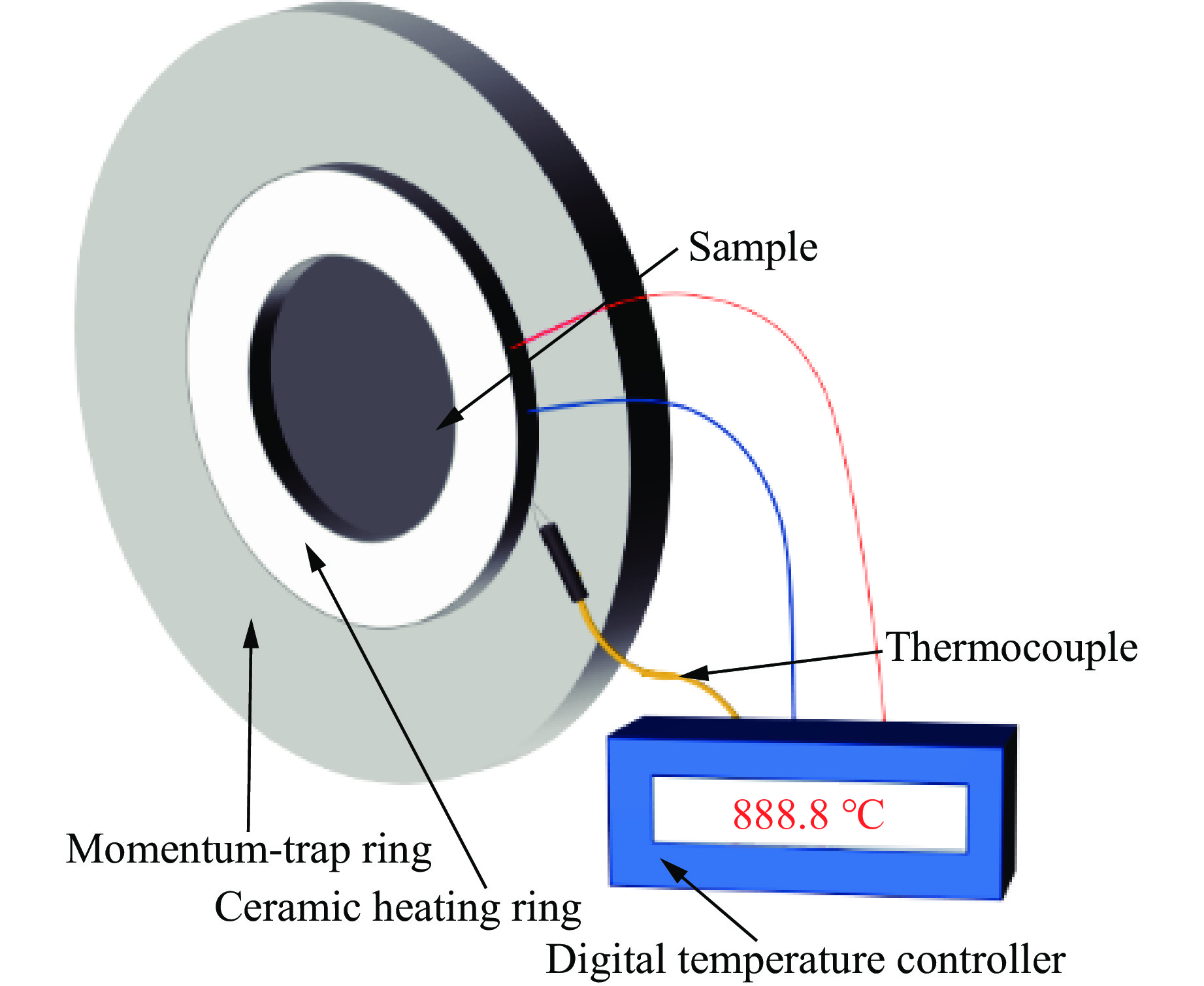
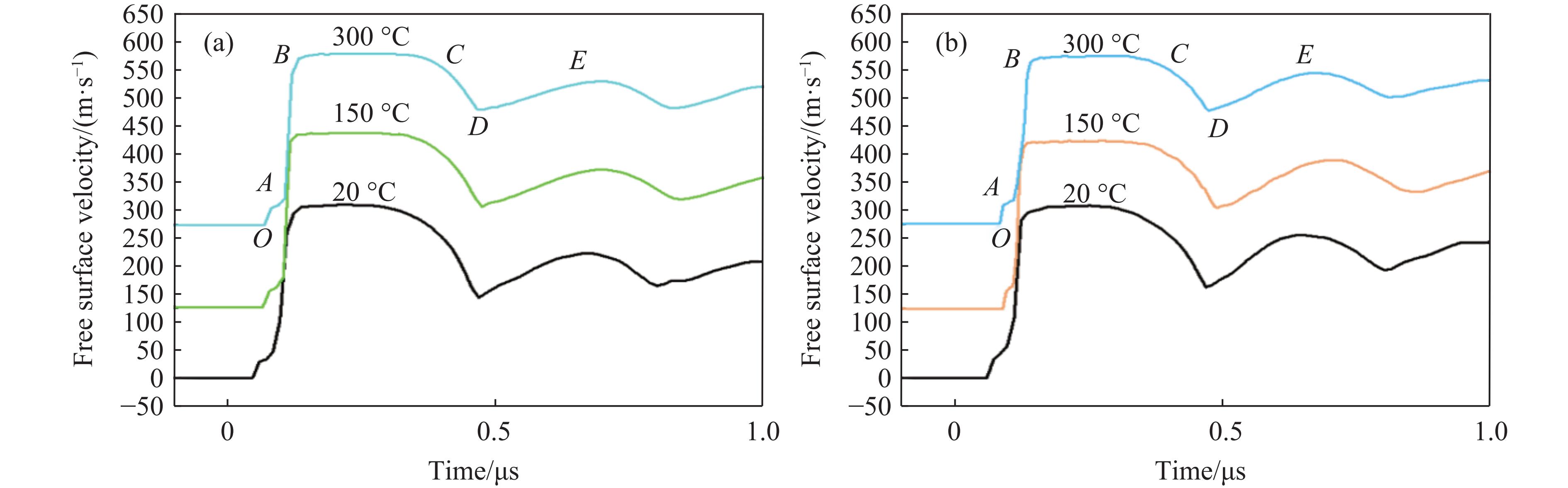
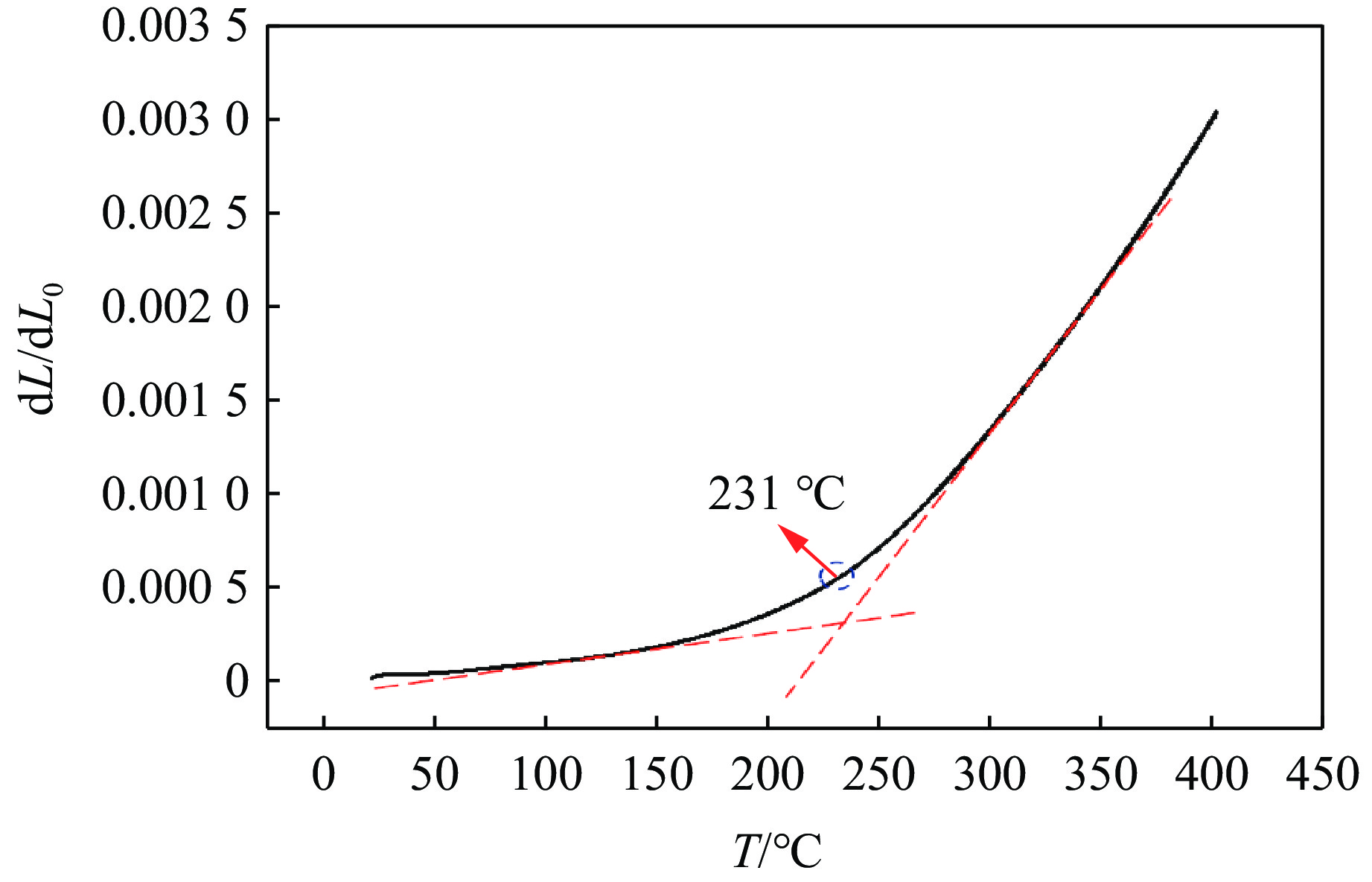
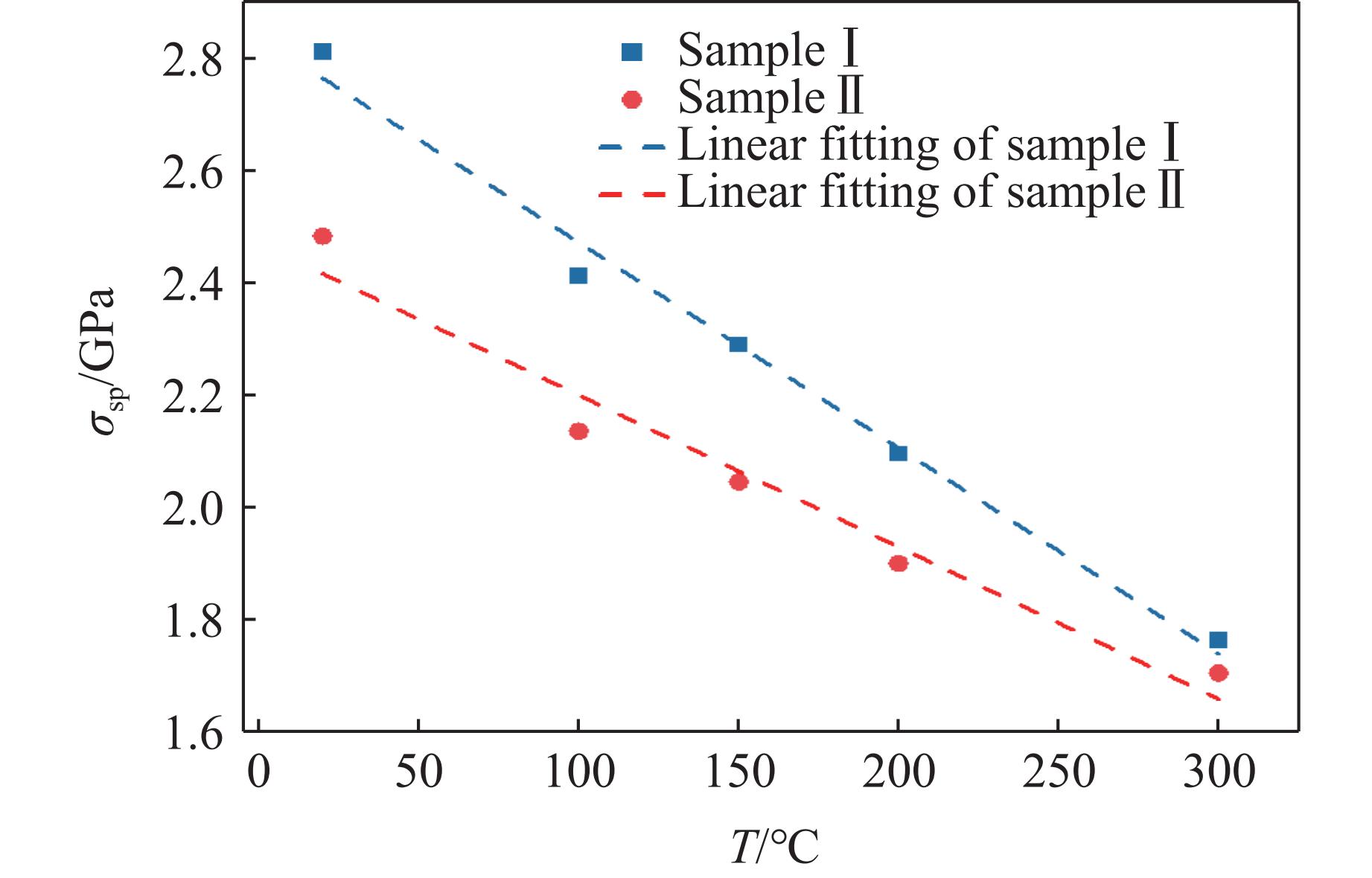
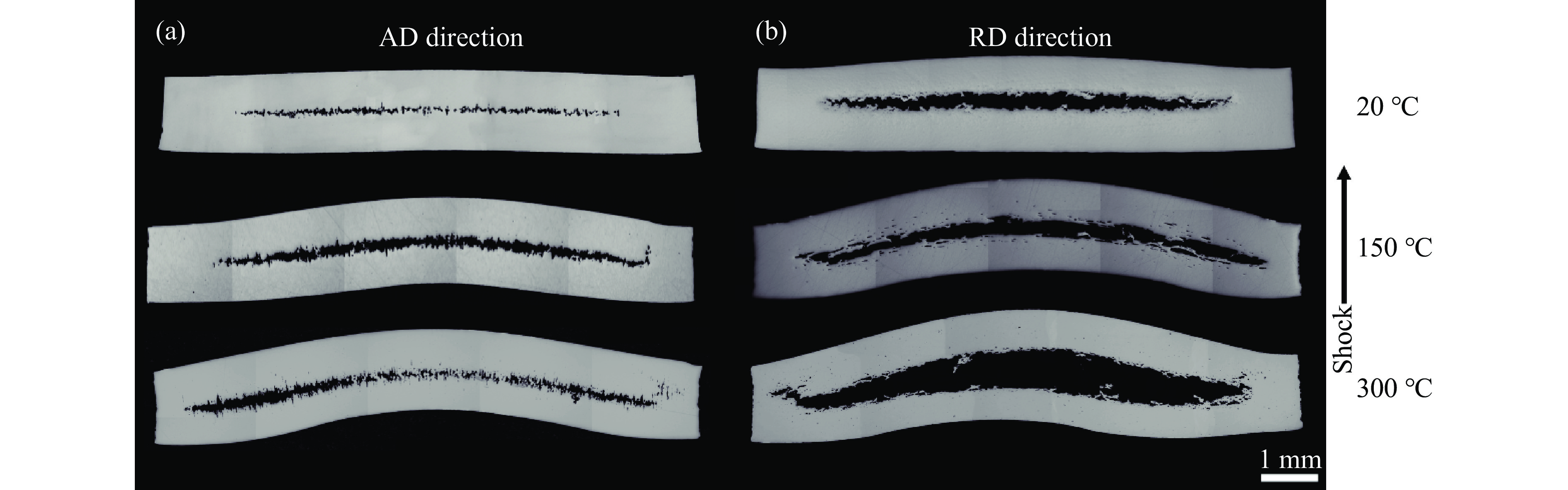
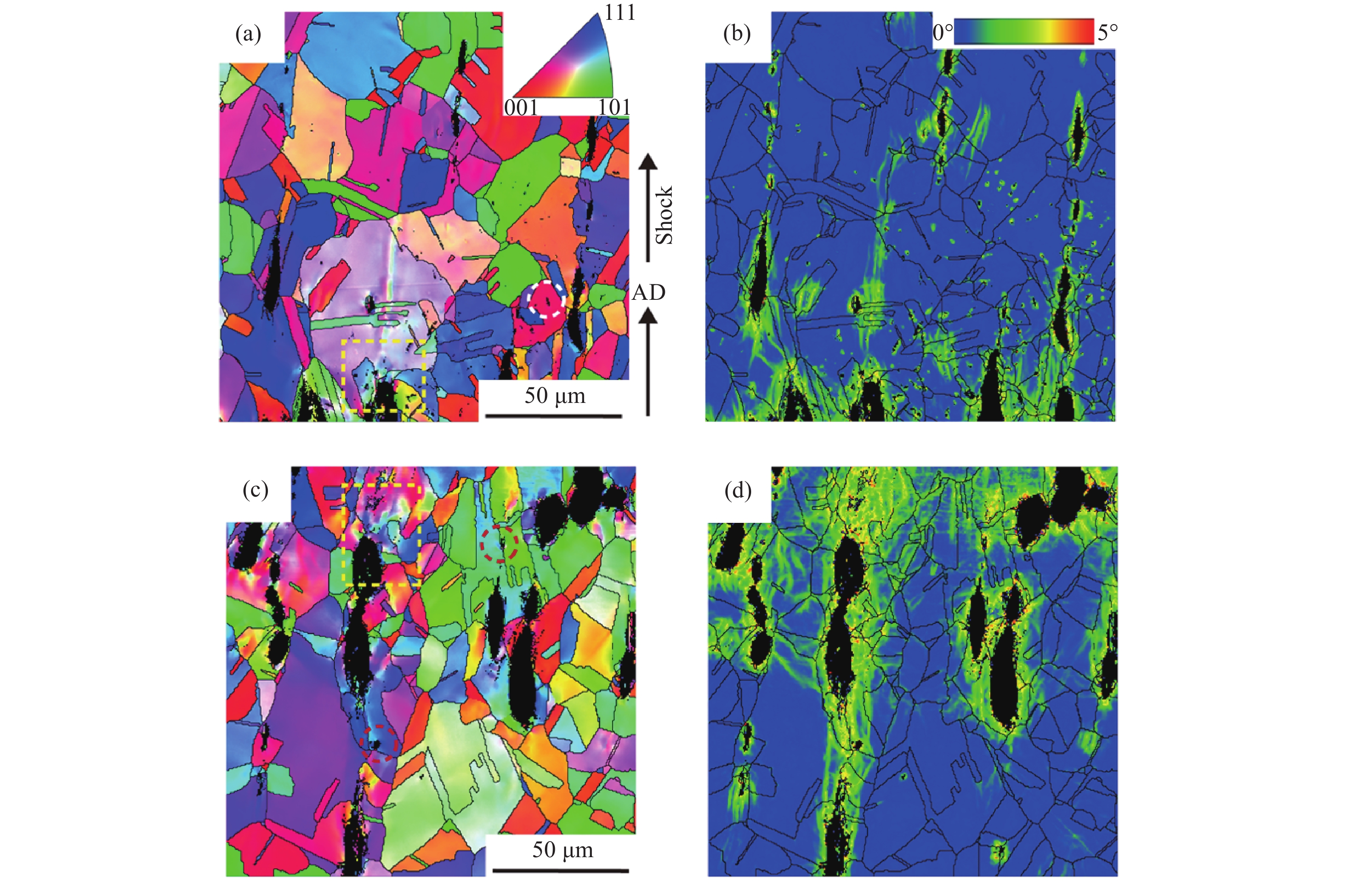

2026,
40(8): 080108.
doi: 10.11858/gywlxb.20261052
Abstract:
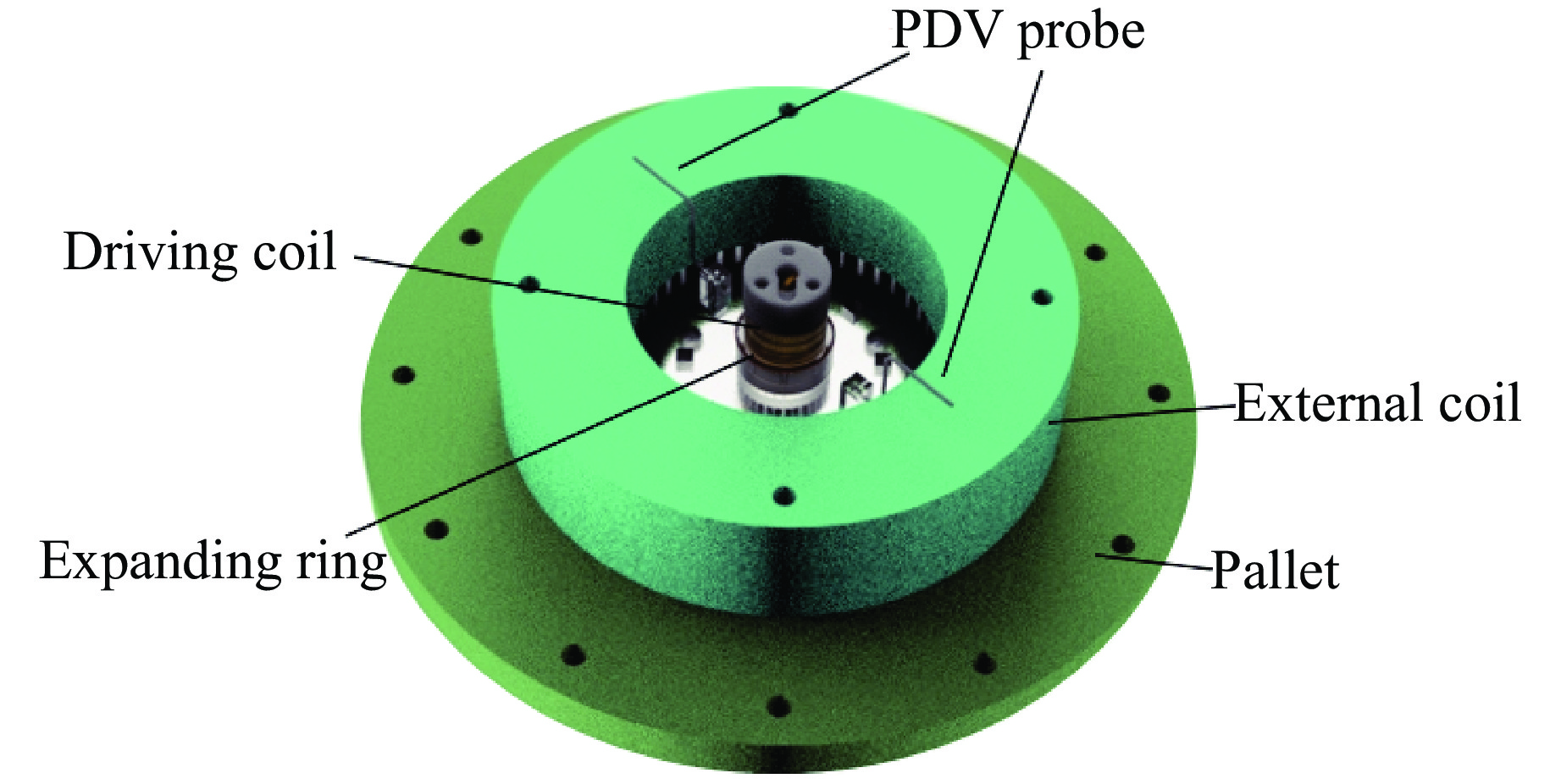
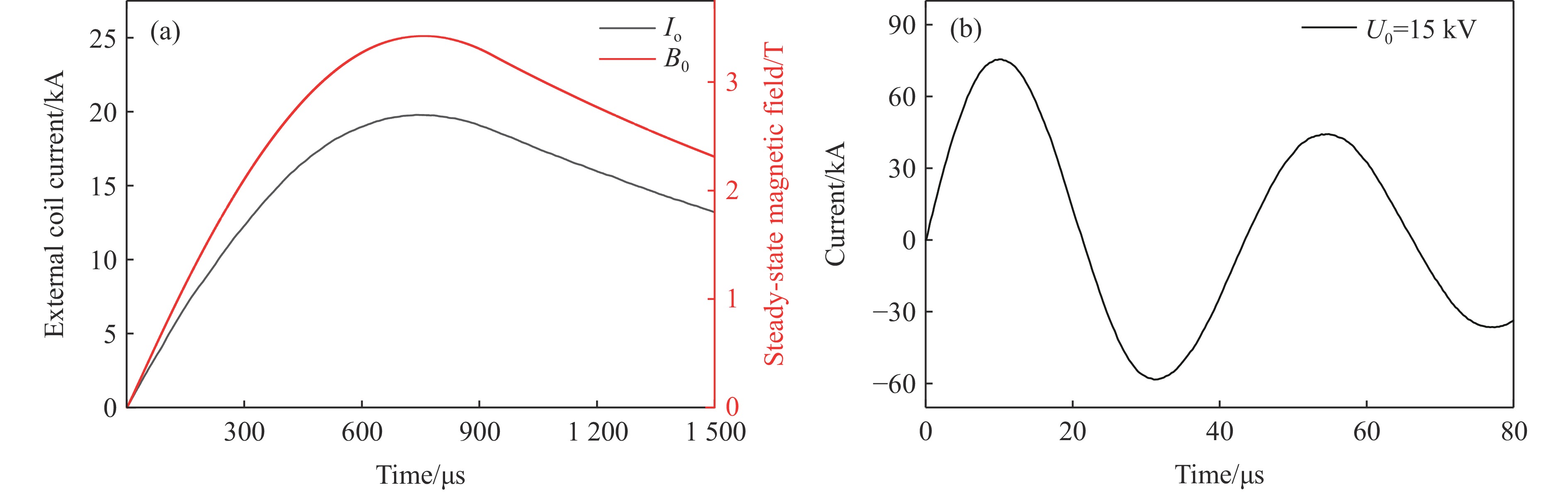

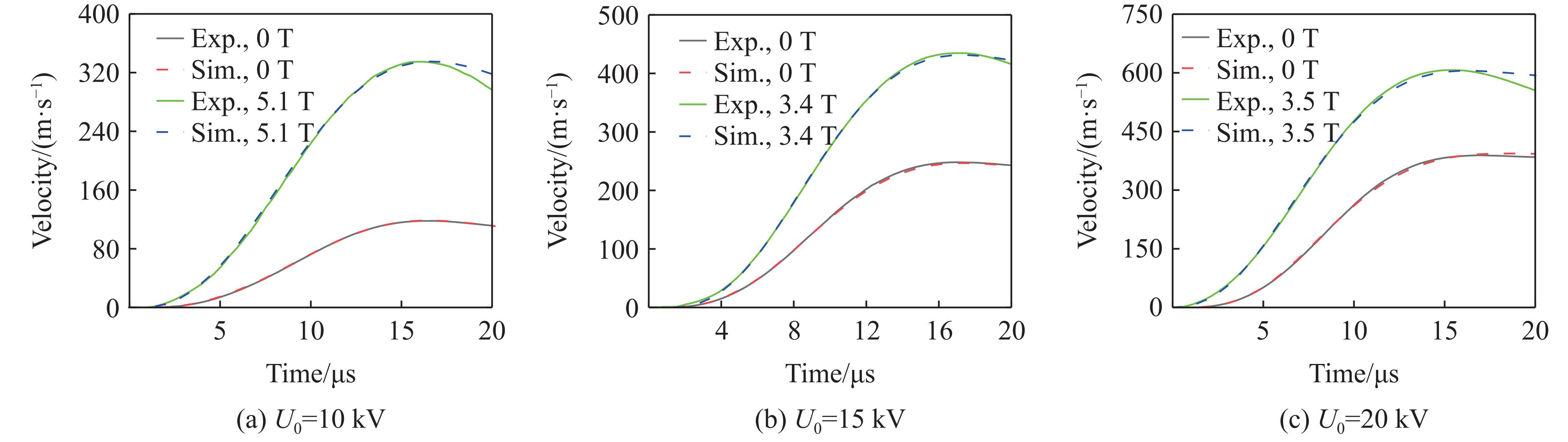
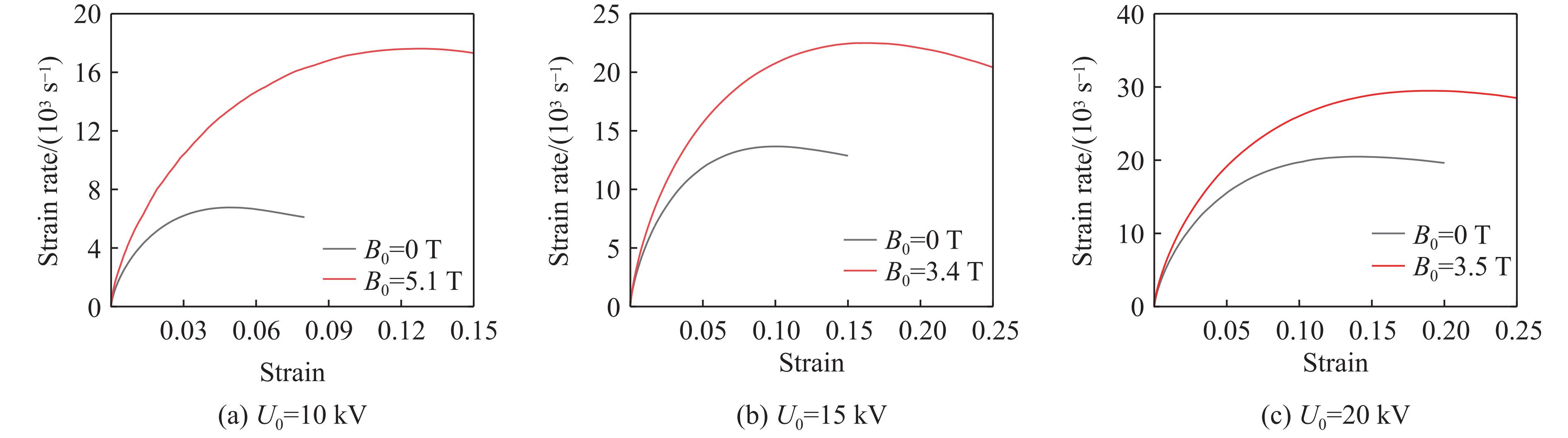
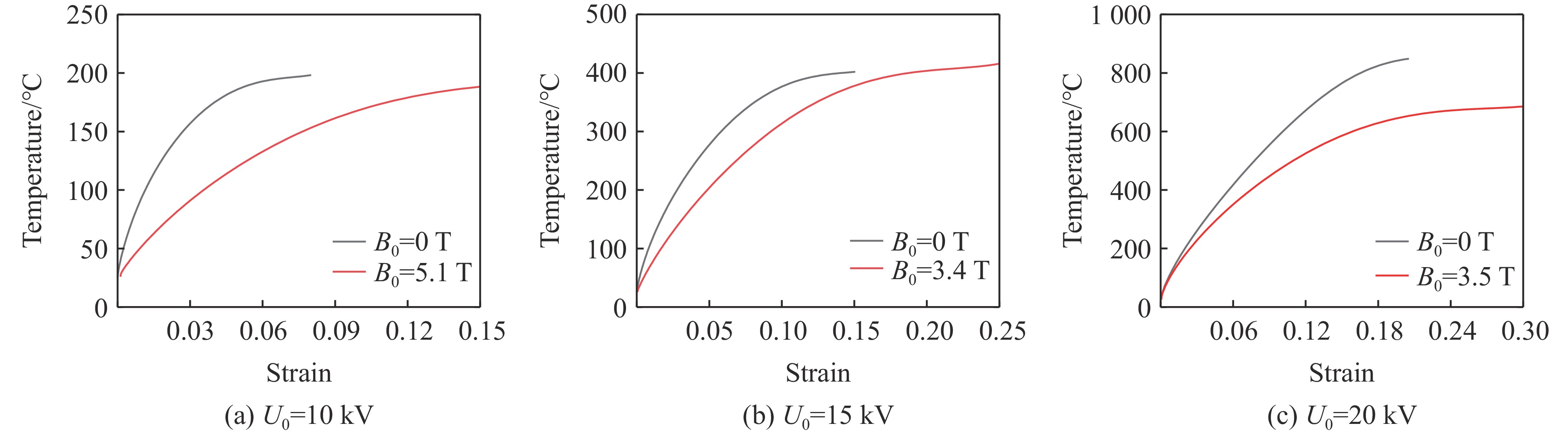
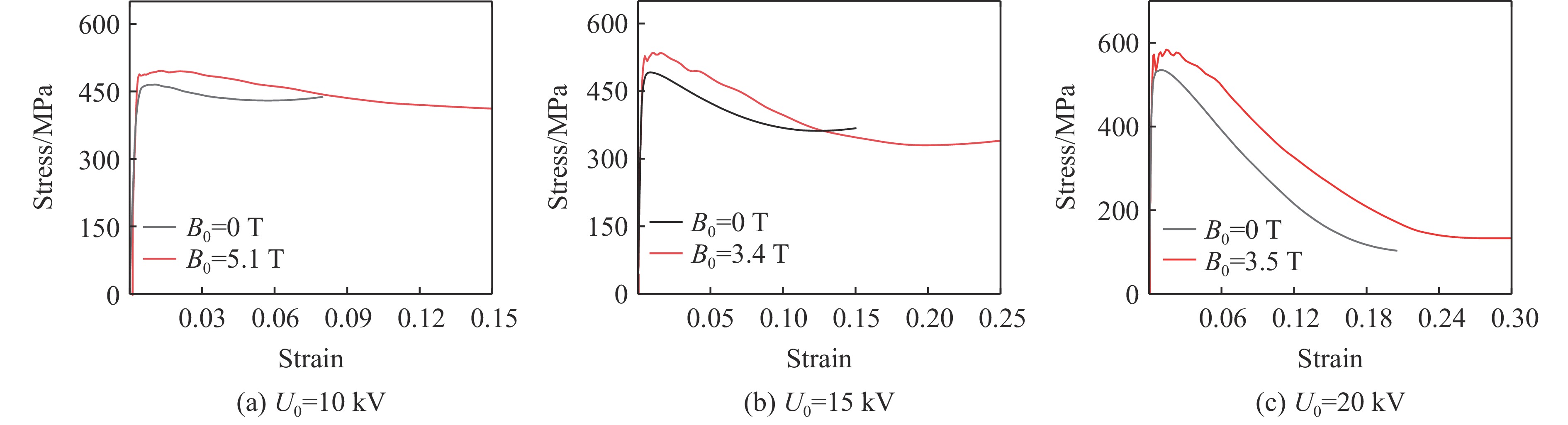
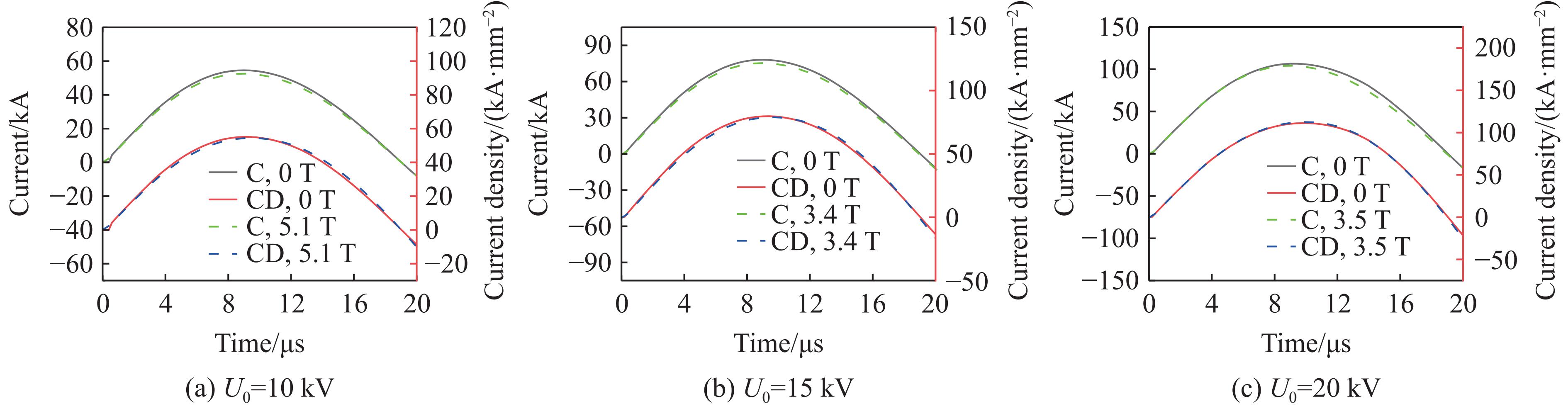
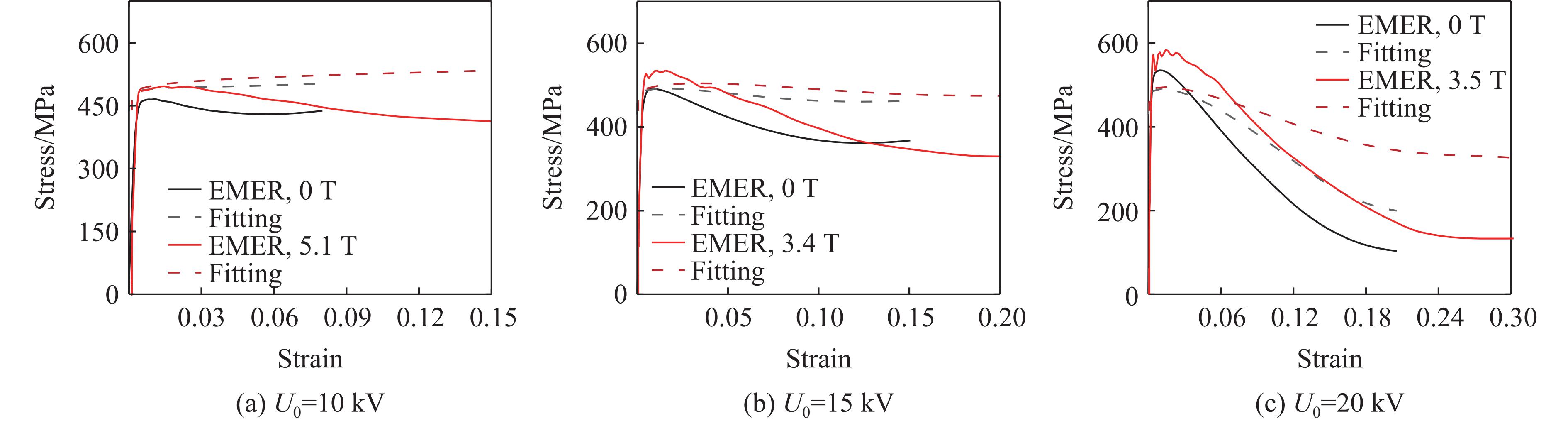
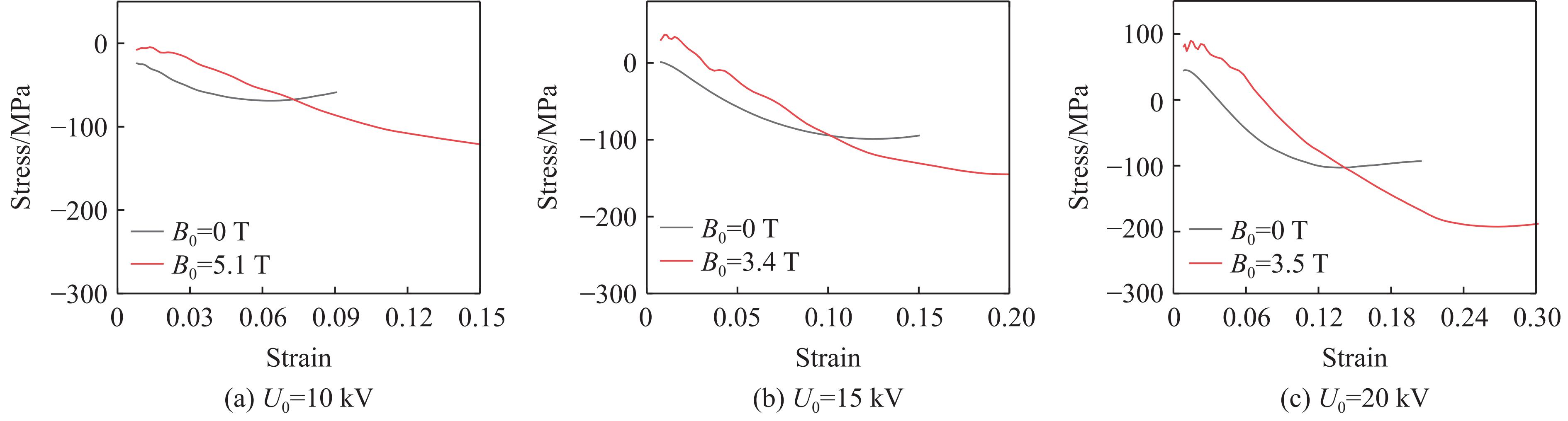
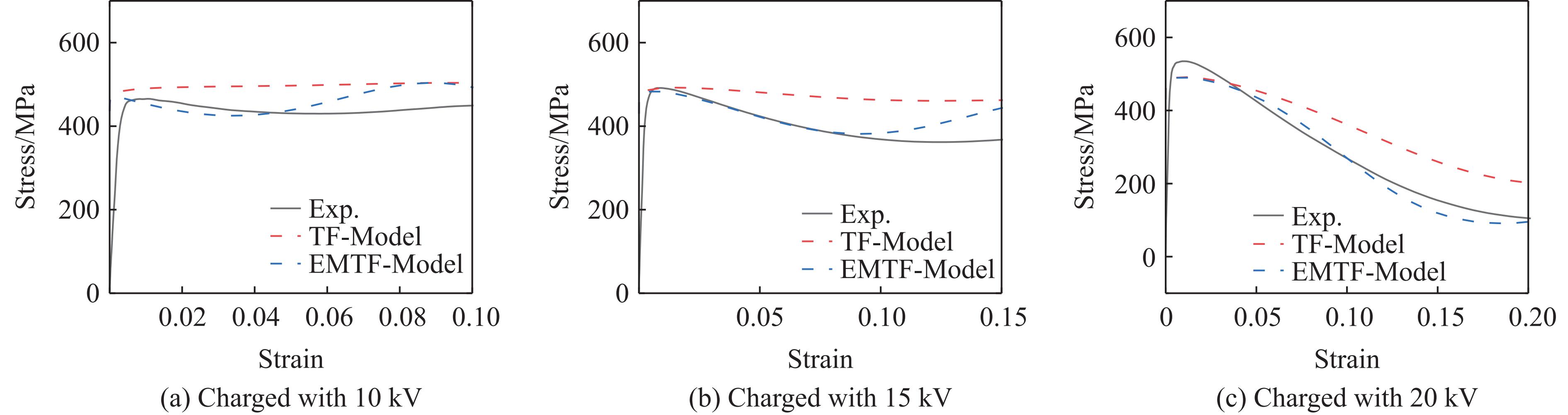
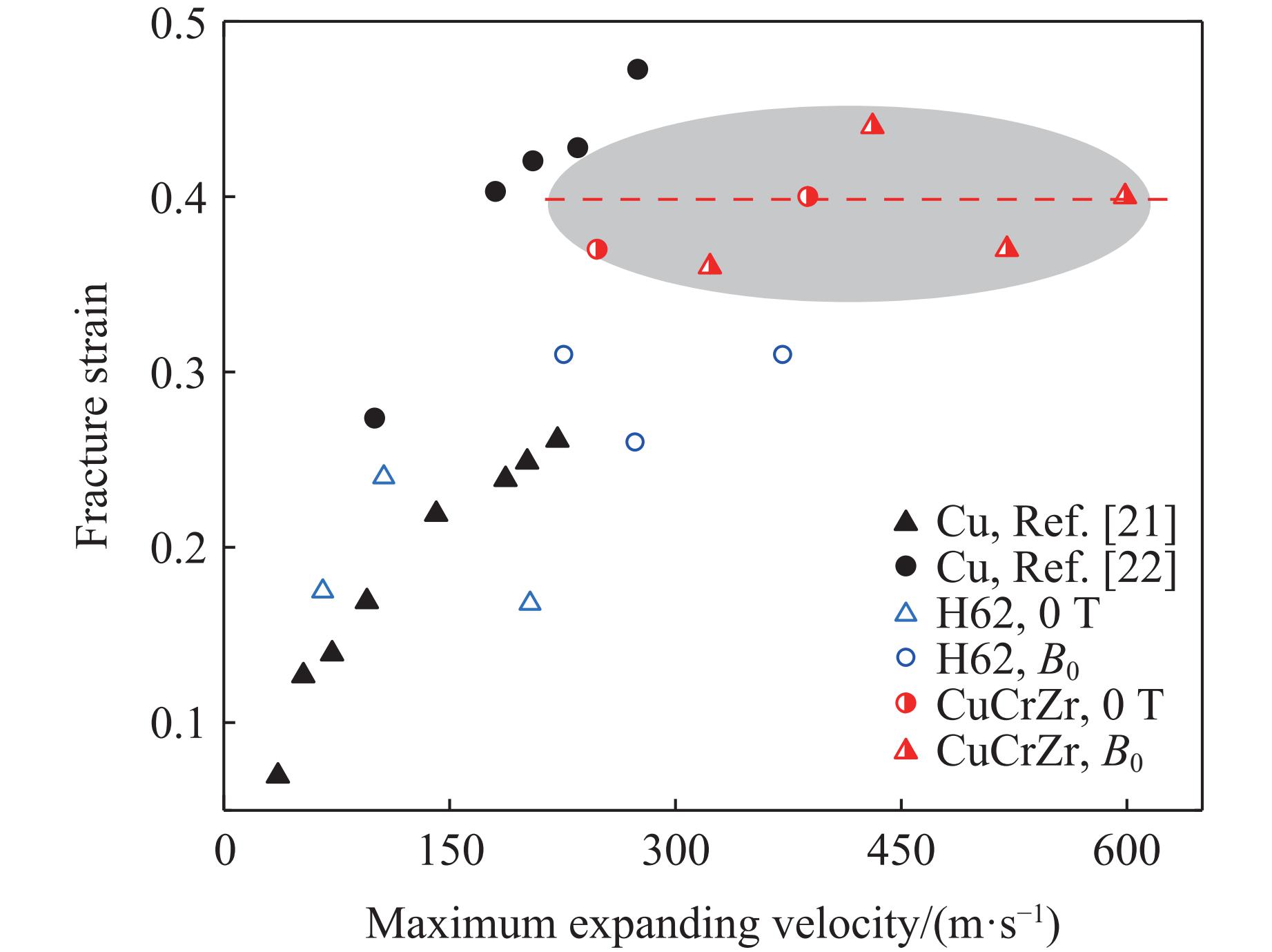


2026,
40(8): 080109.
doi: 10.11858/gywlxb.20251264
Abstract:
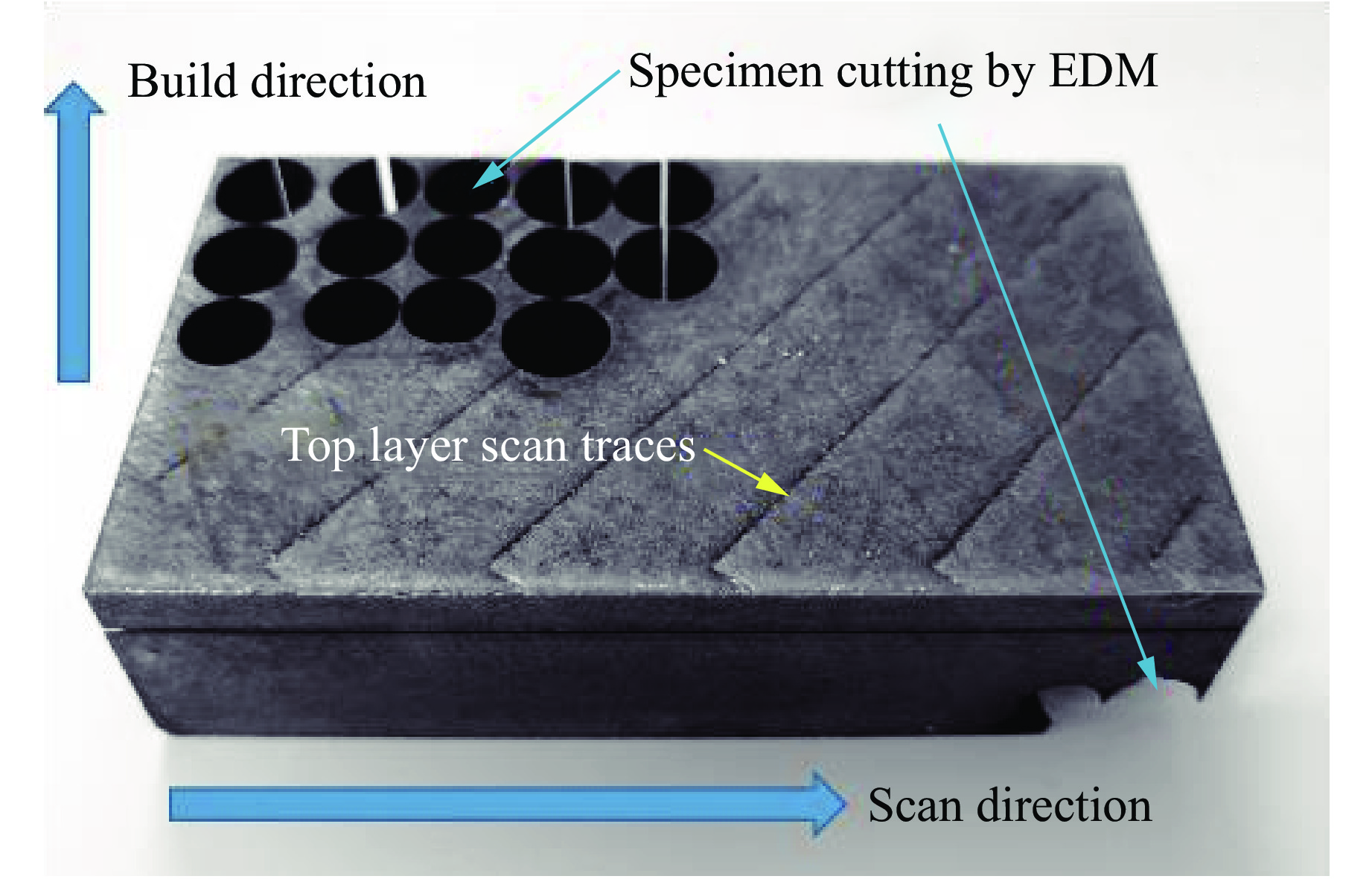
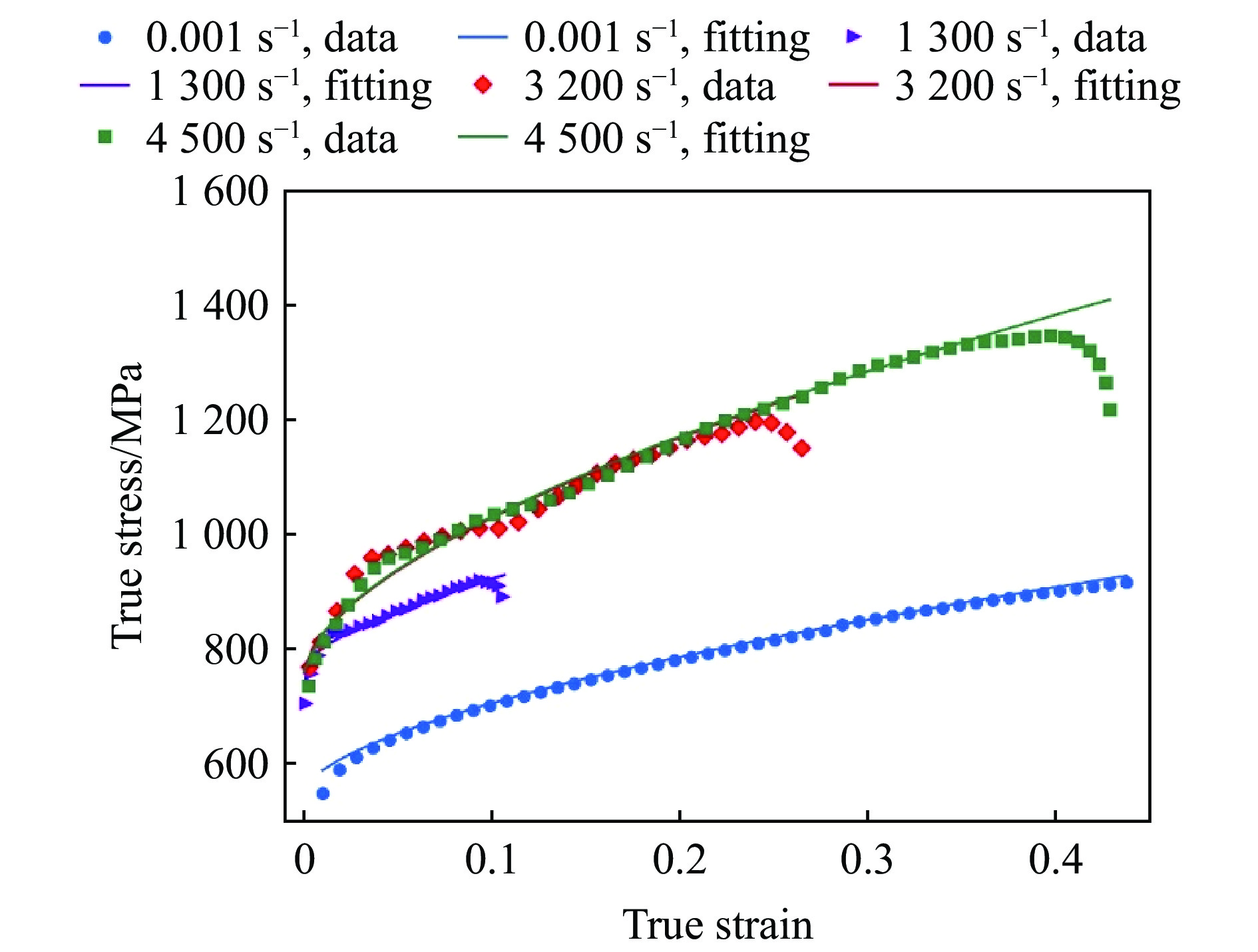
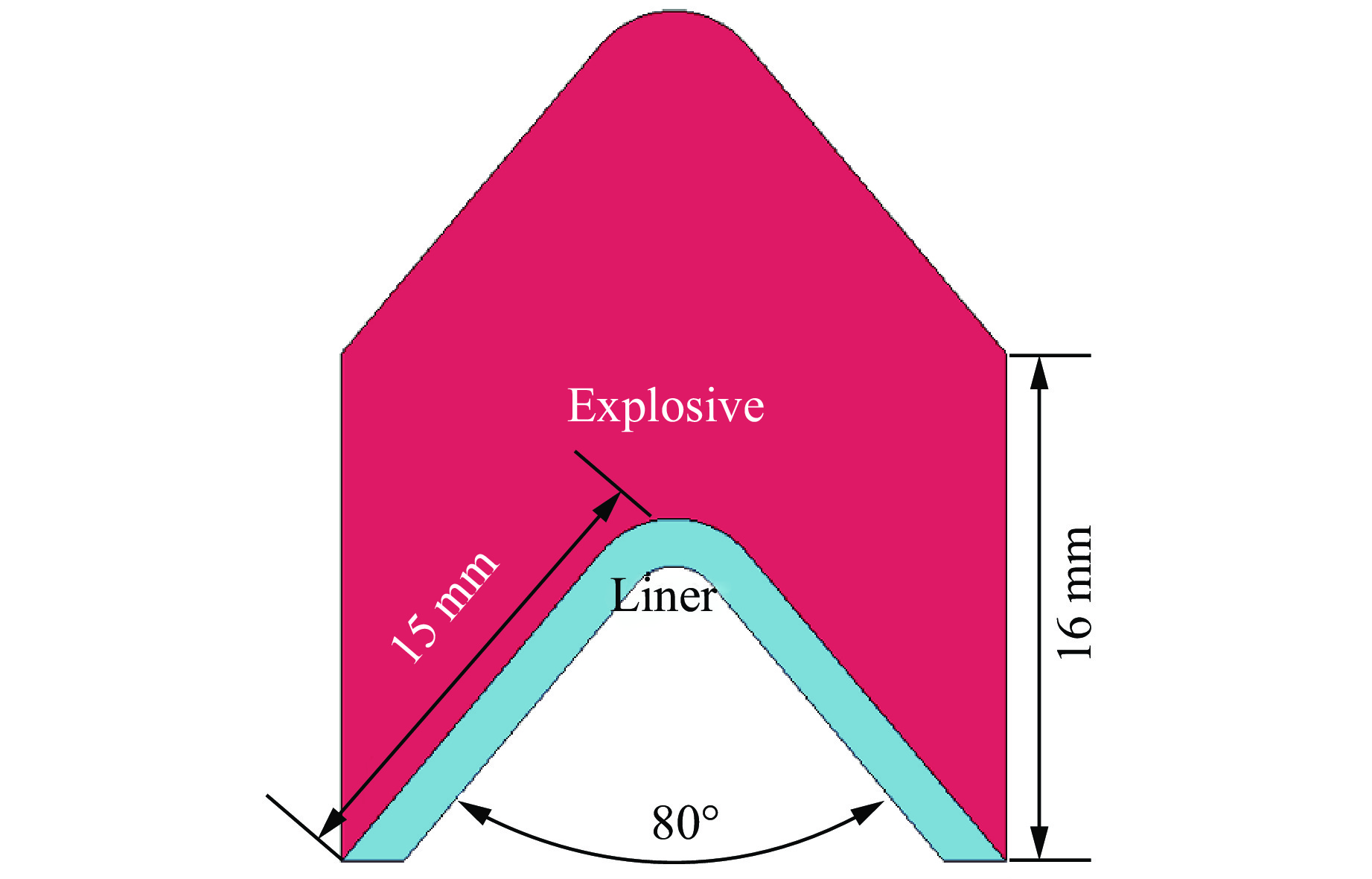
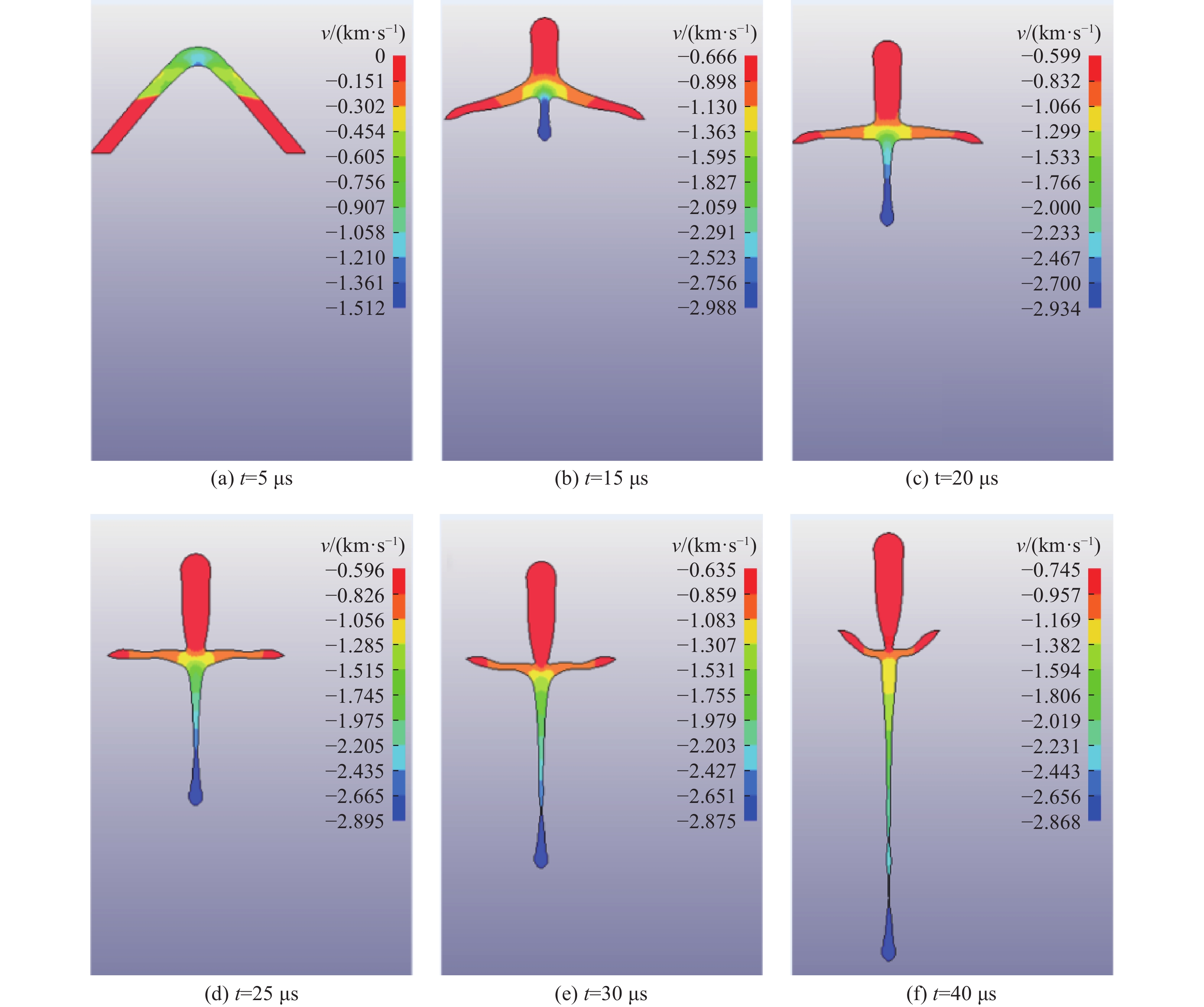
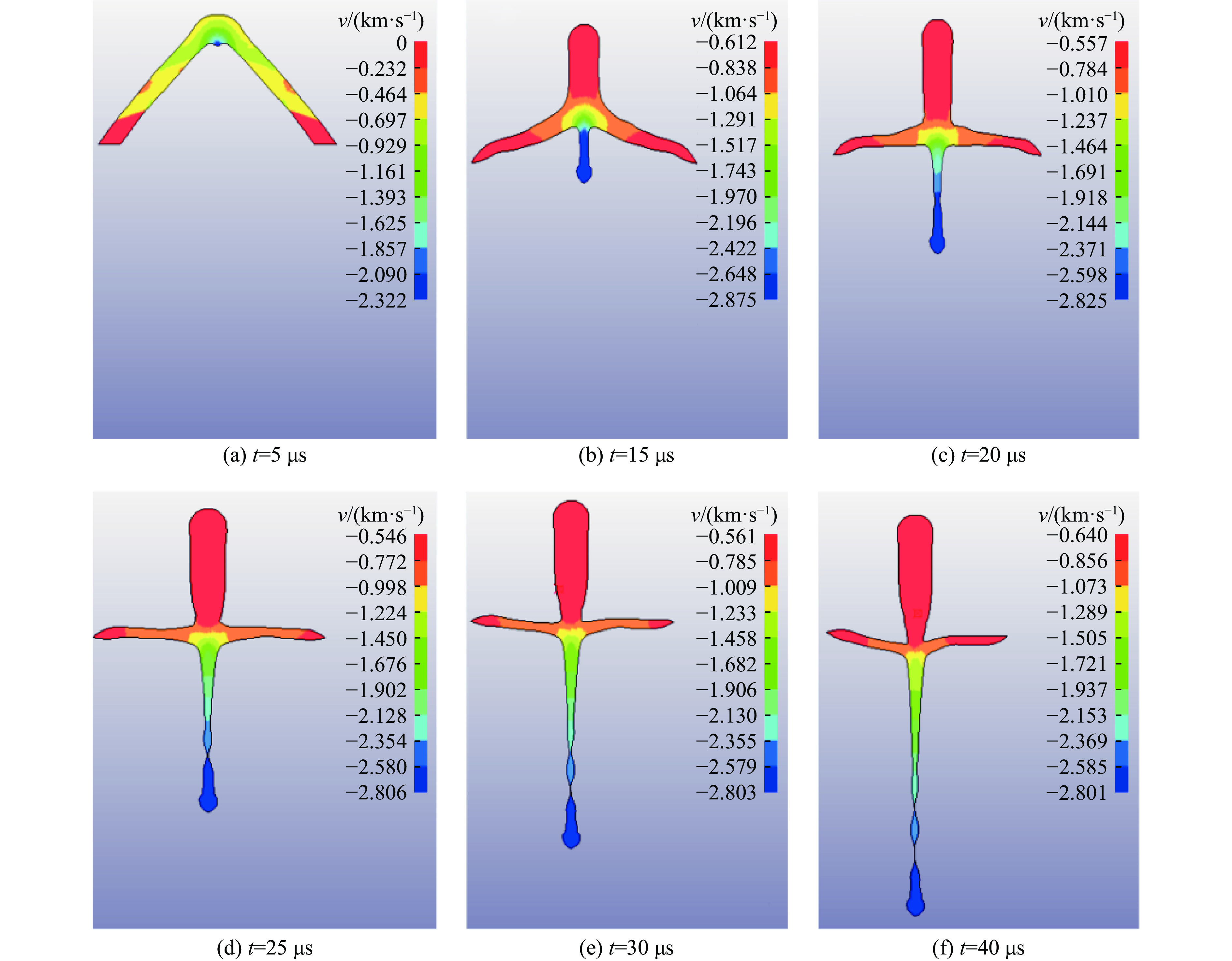
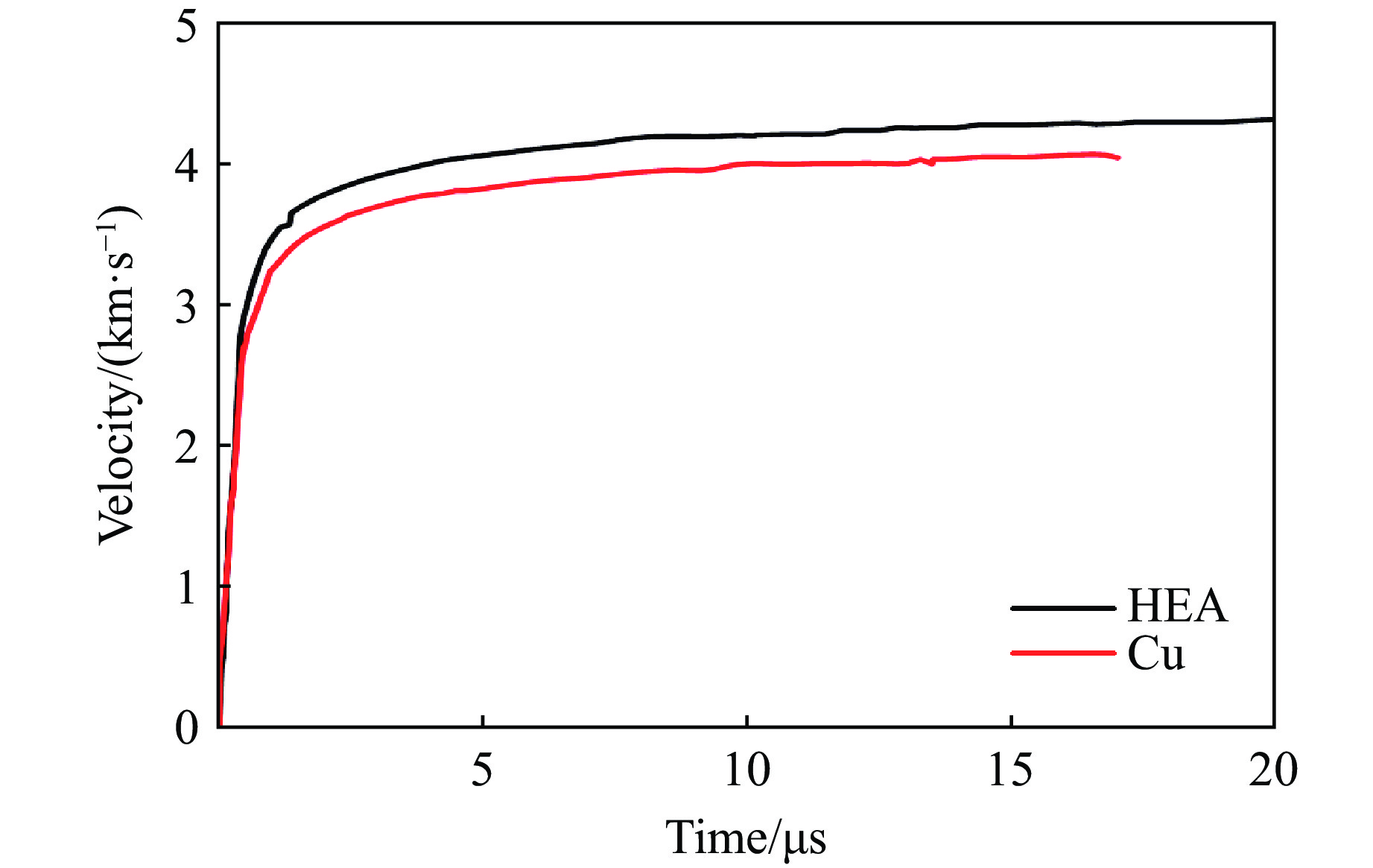
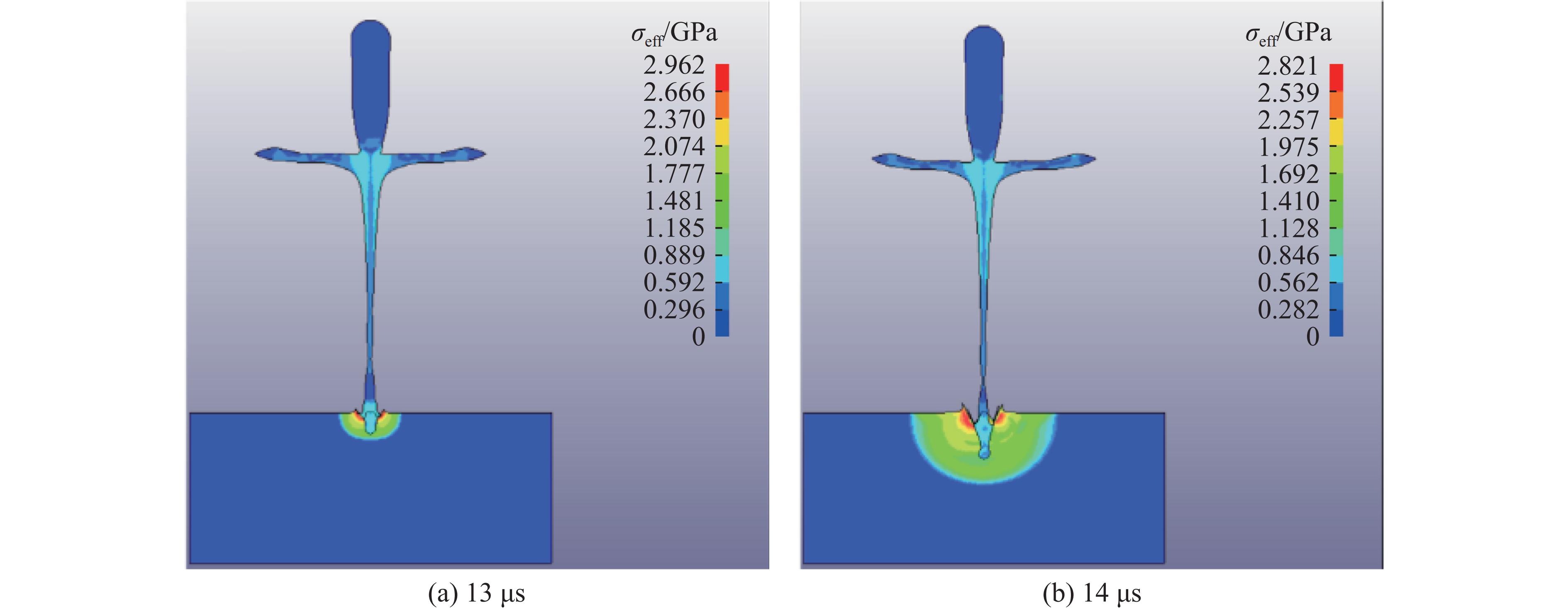
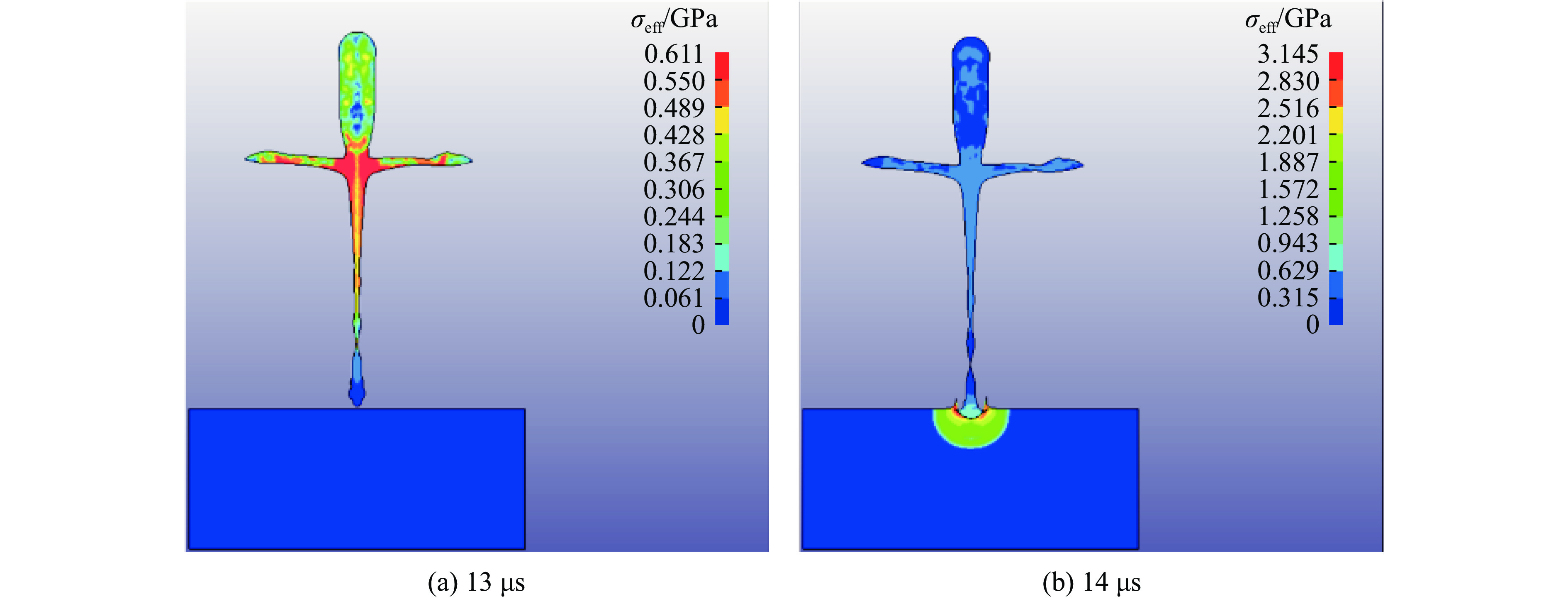
2026,
40(8): 080110.
doi: 10.11858/gywlxb.20251246
Abstract:
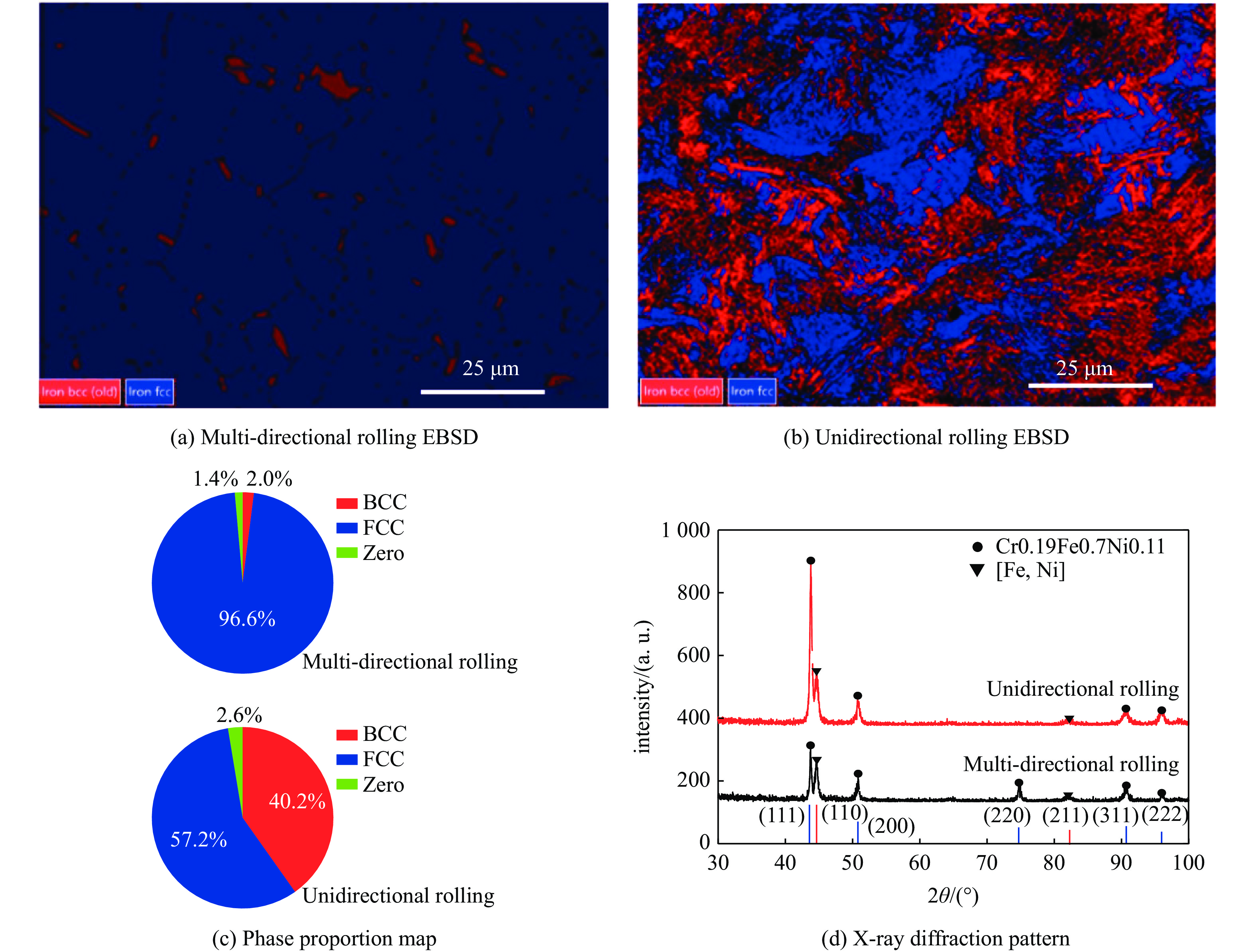
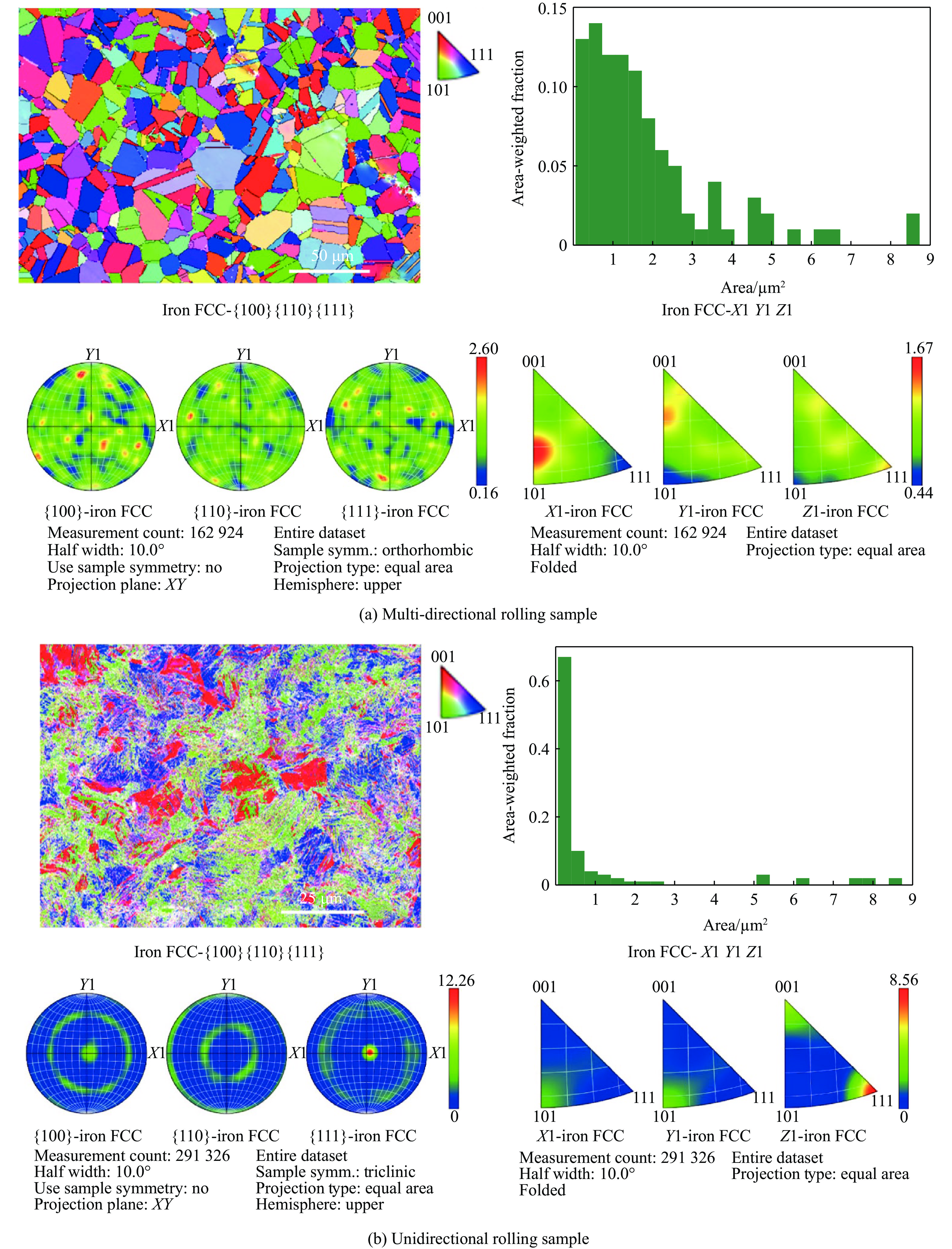
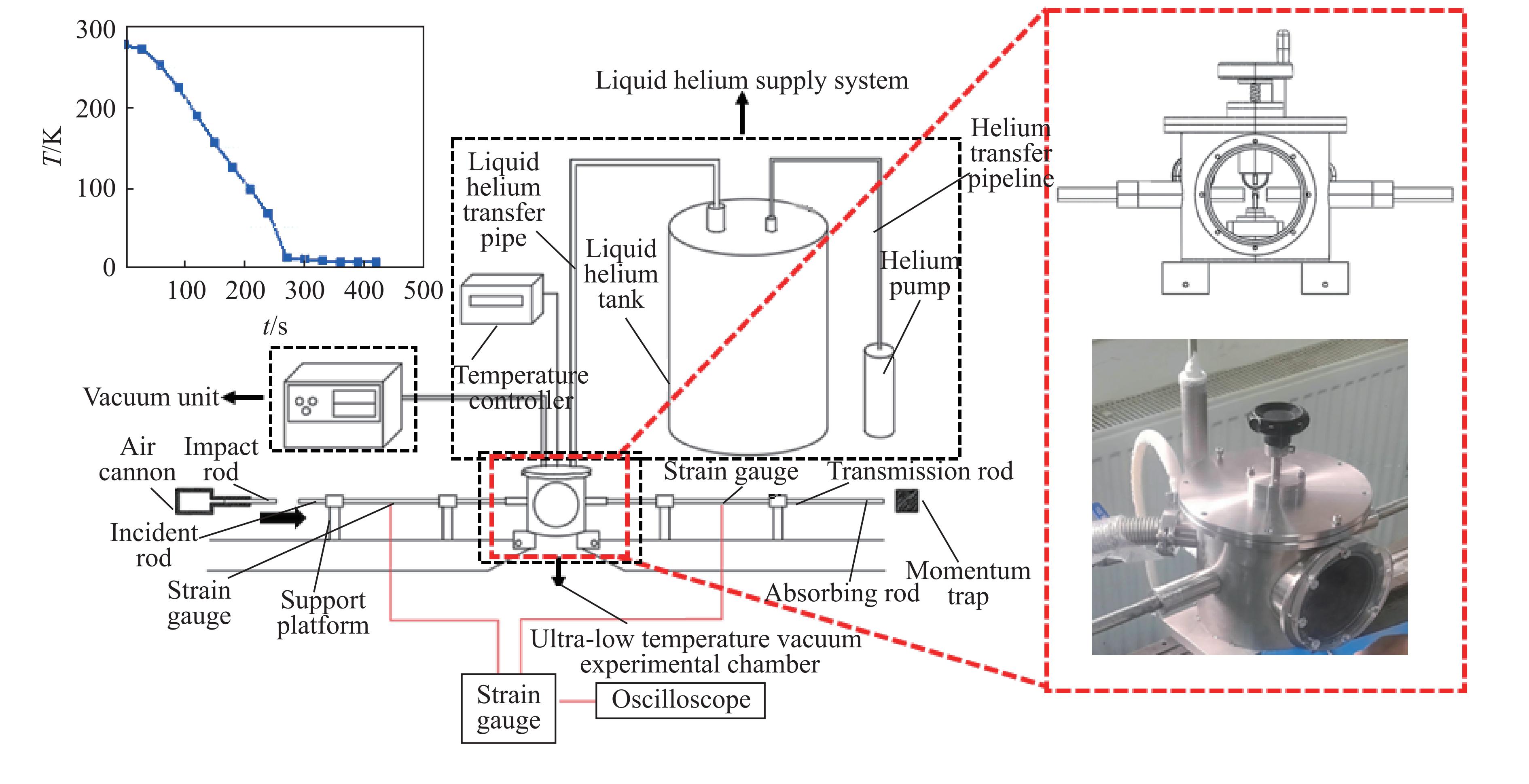
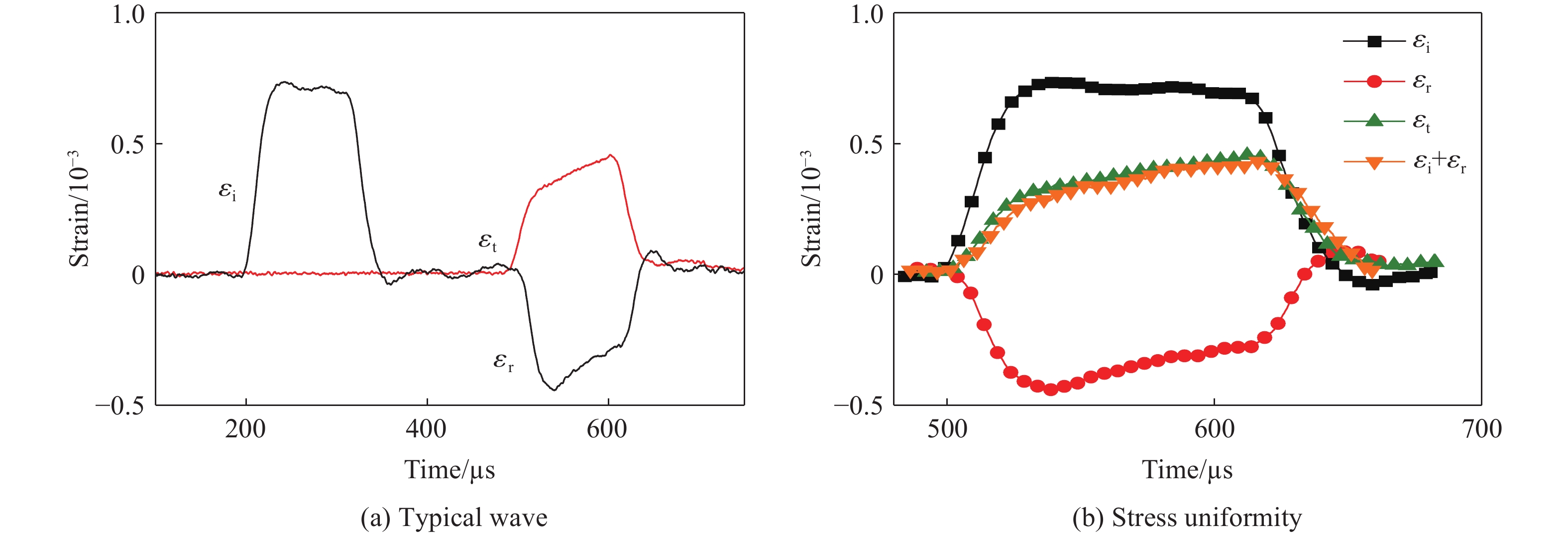
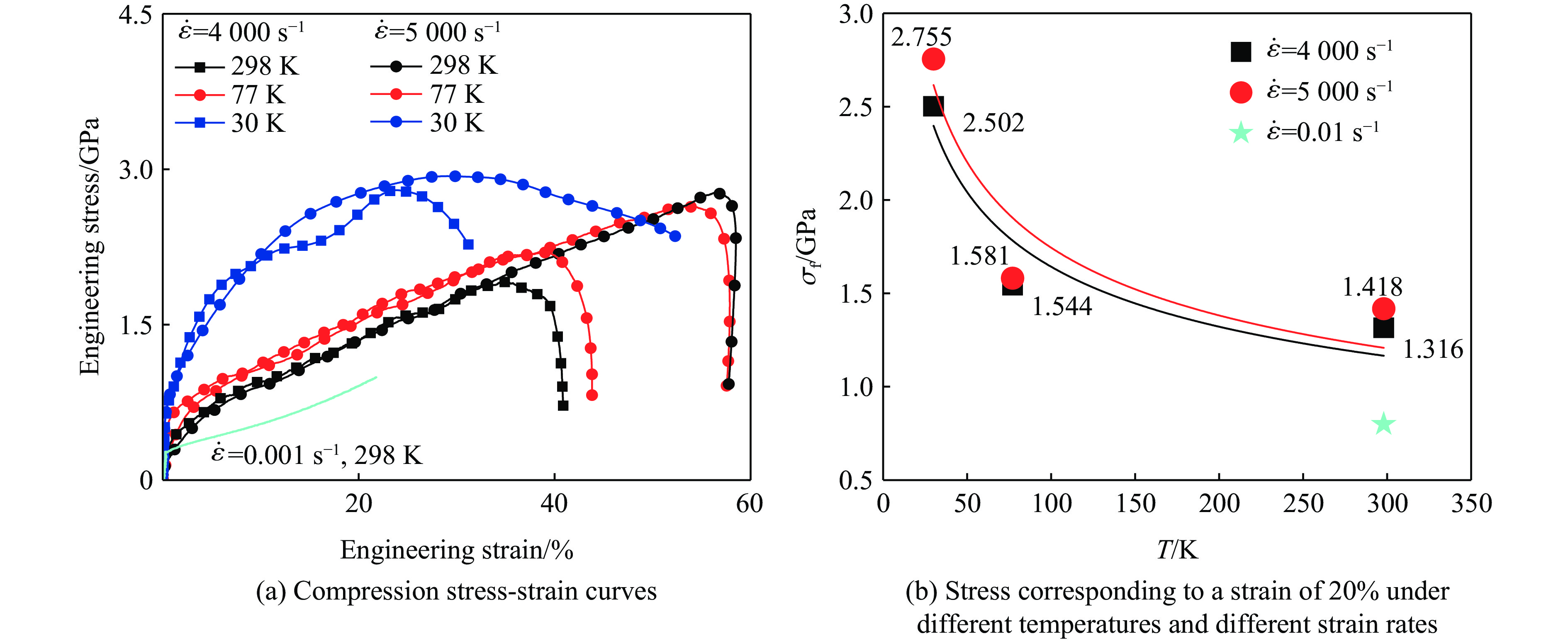
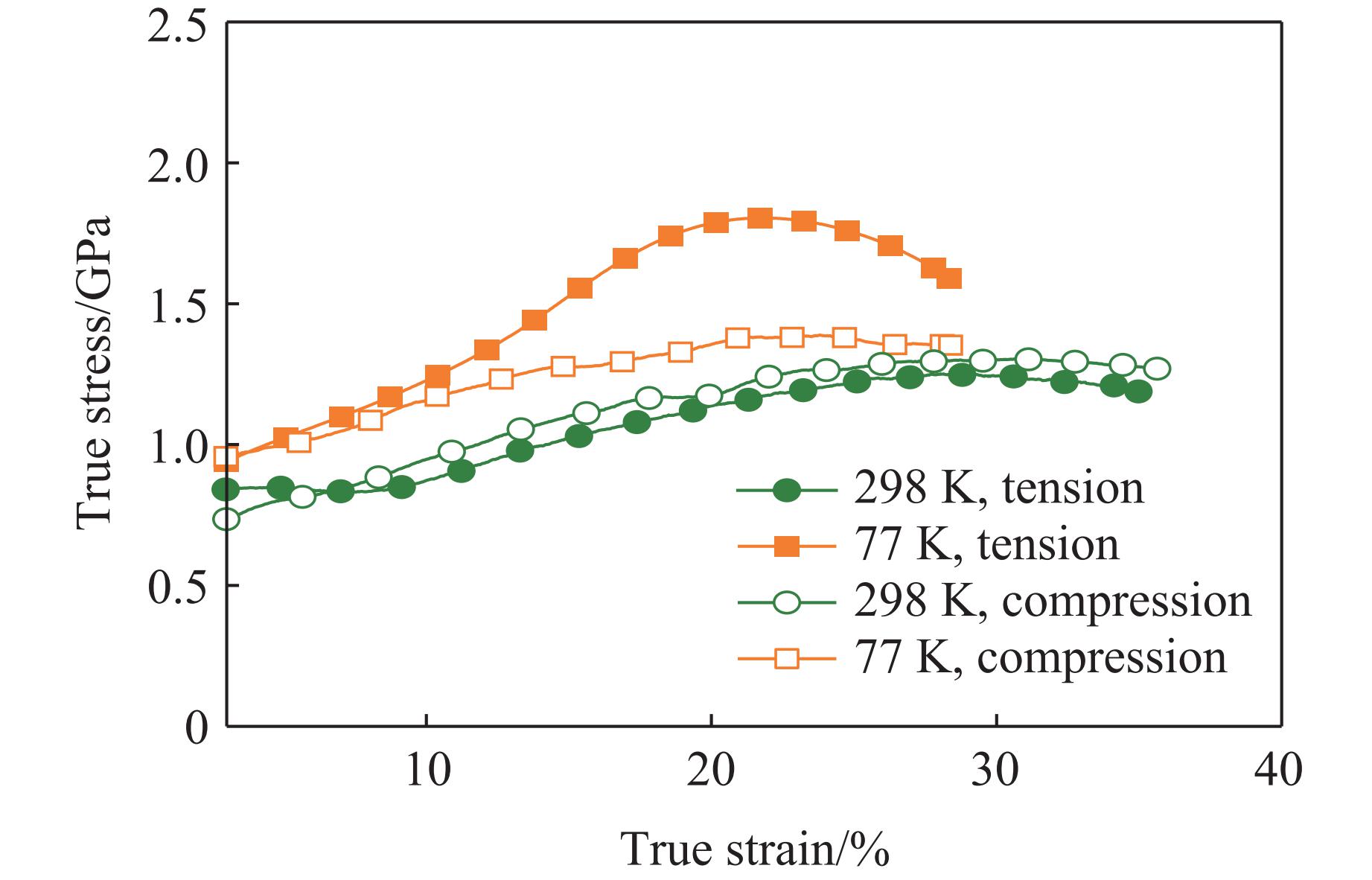
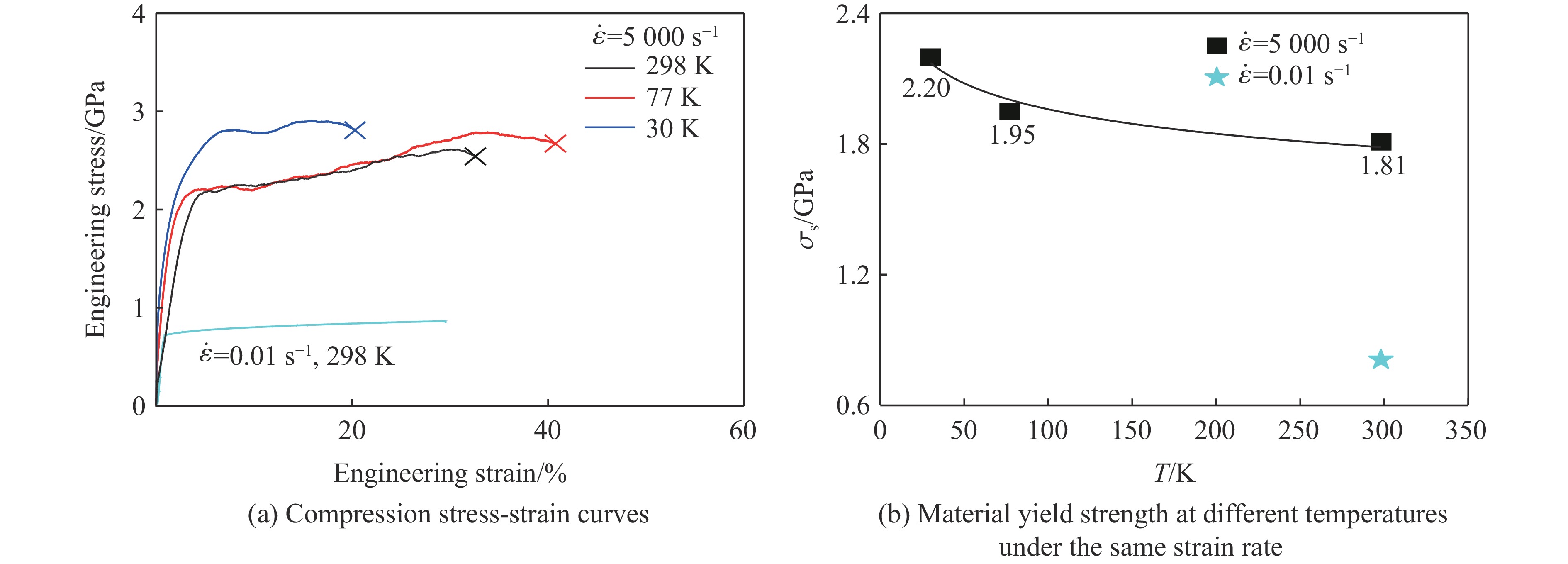
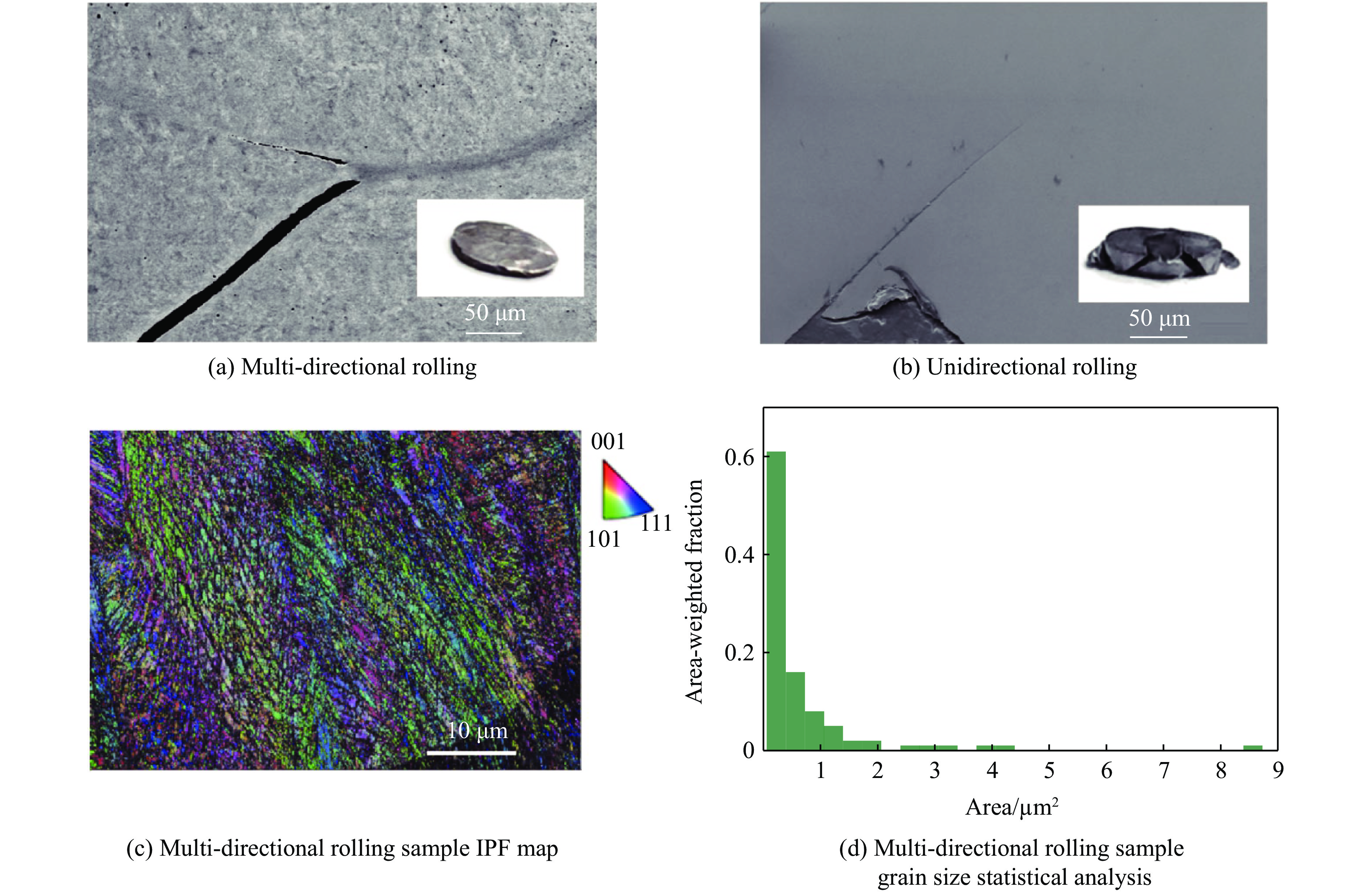
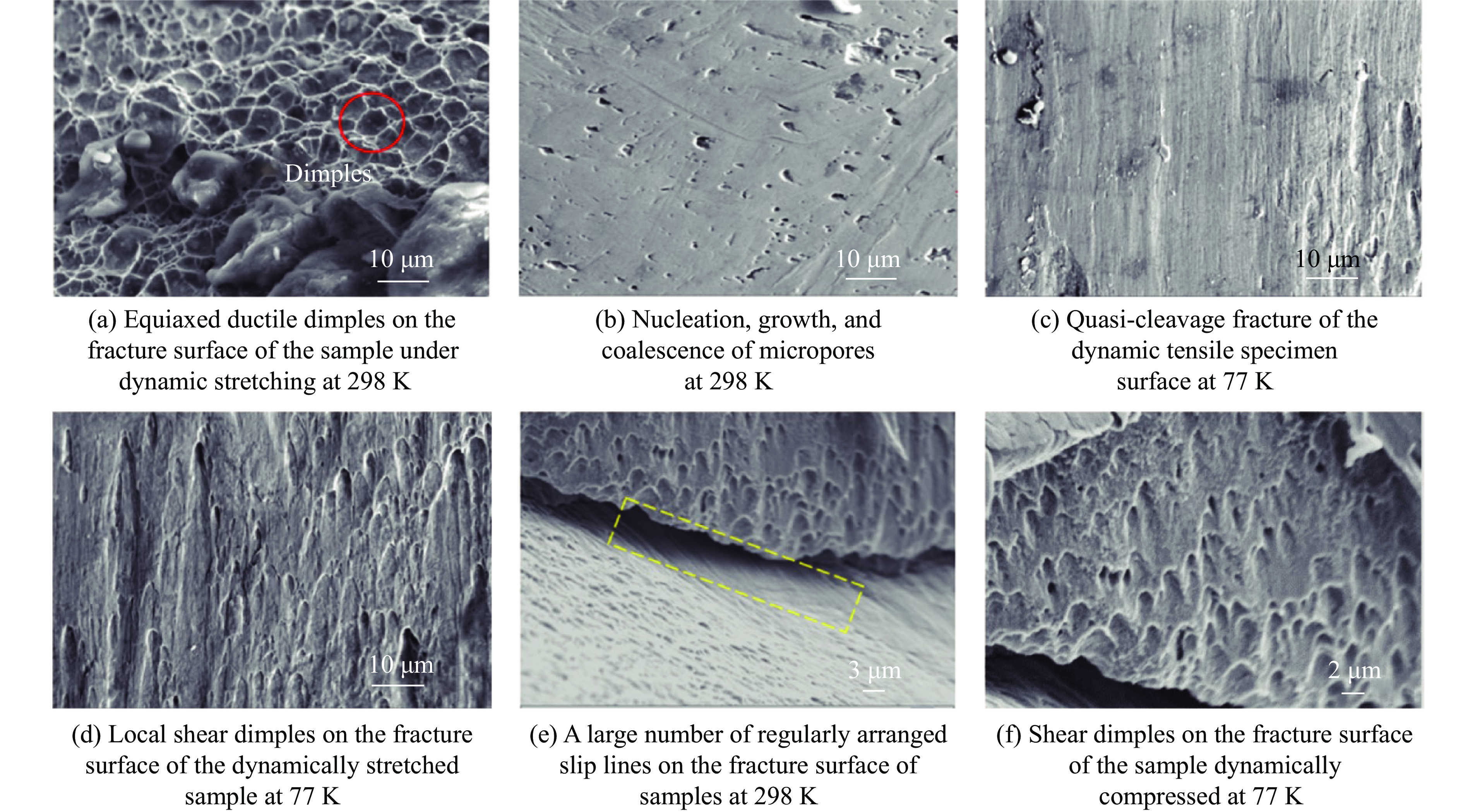
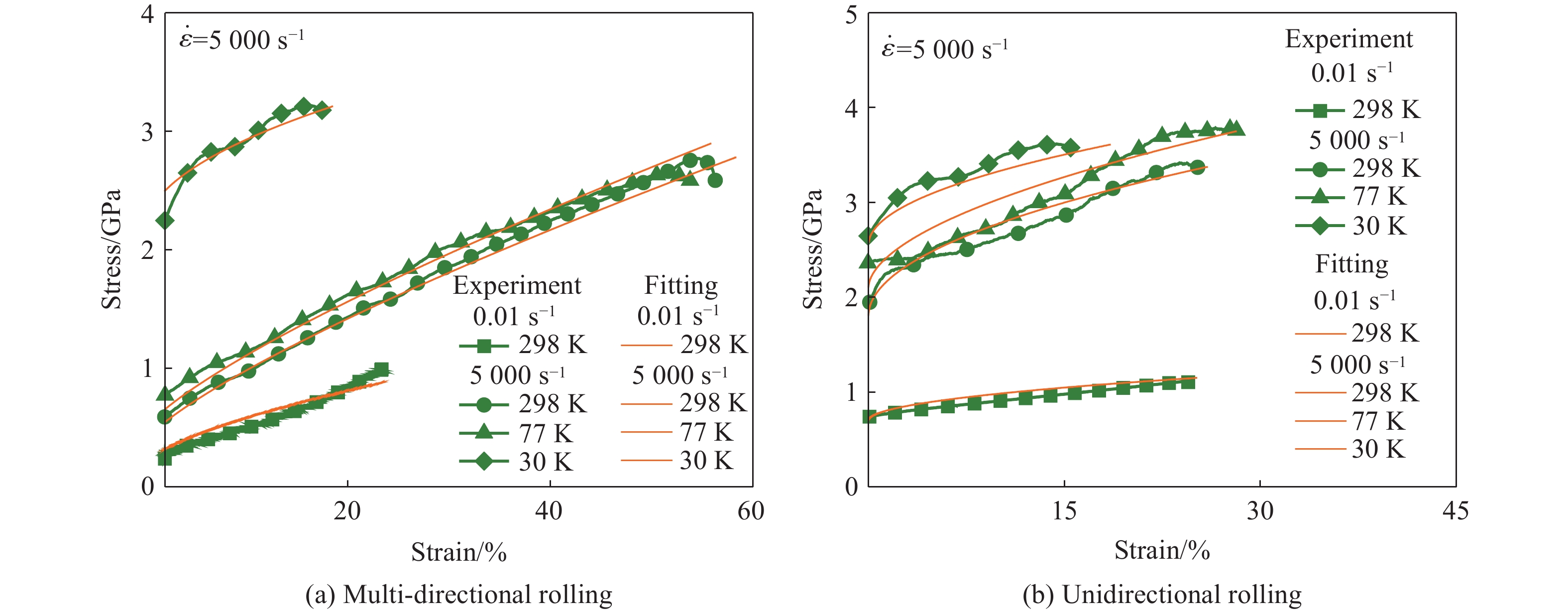
2026,
40(8): 080111.
doi: 10.11858/gywlxb.20251275
Abstract: