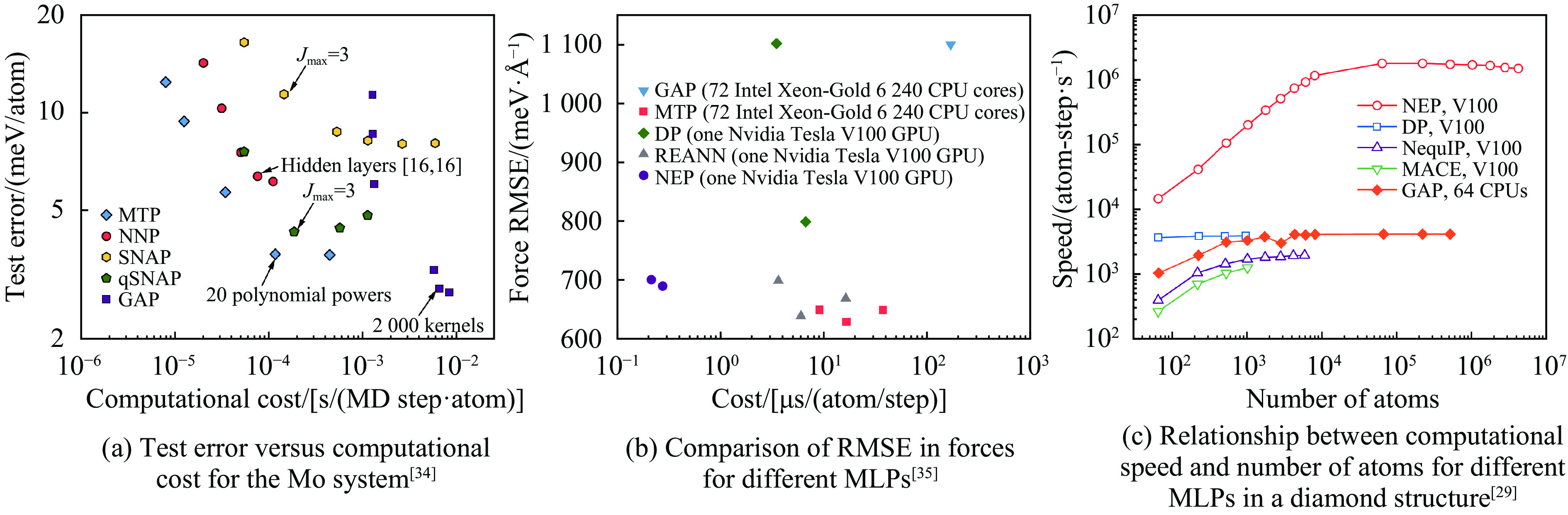
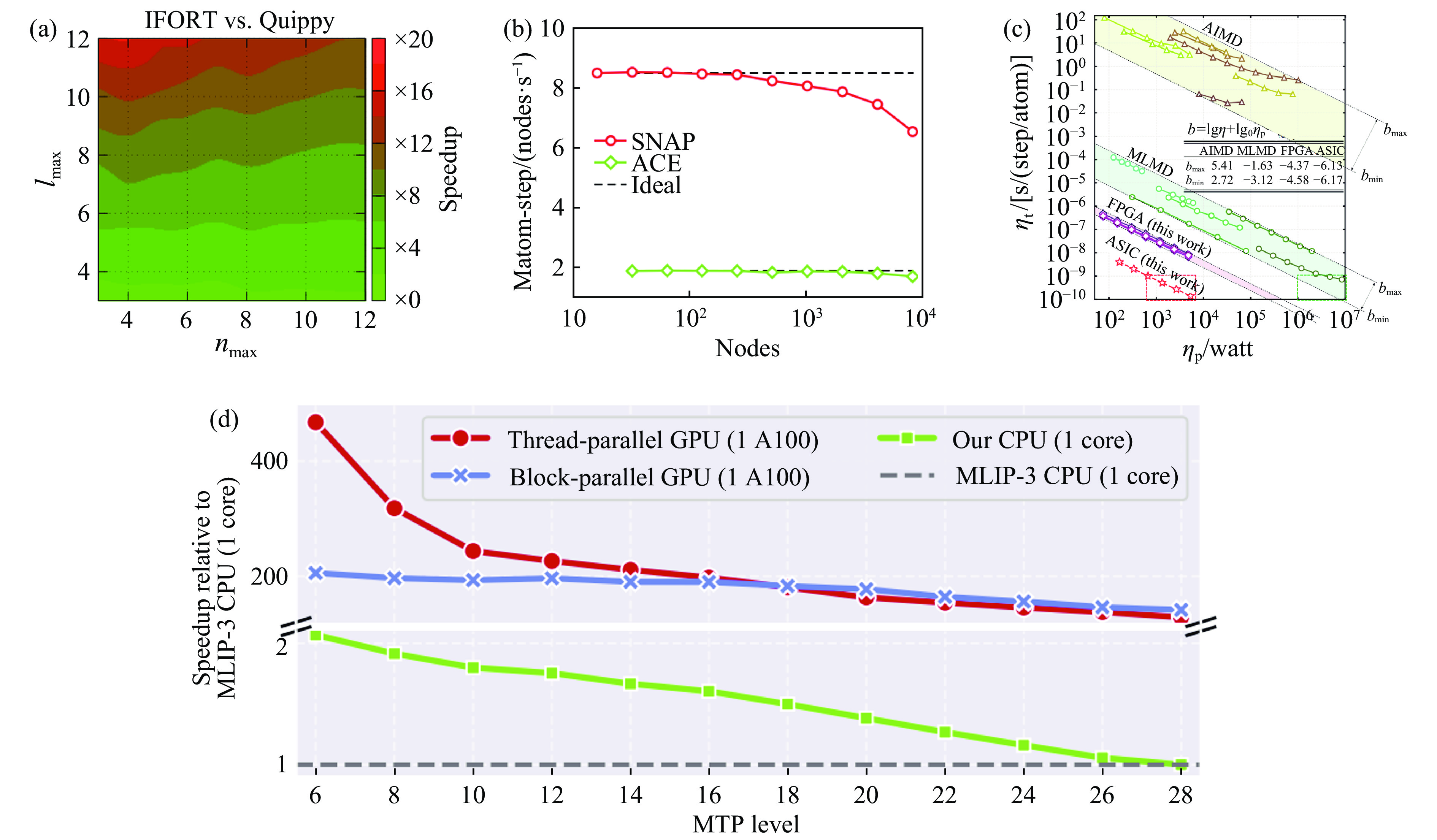
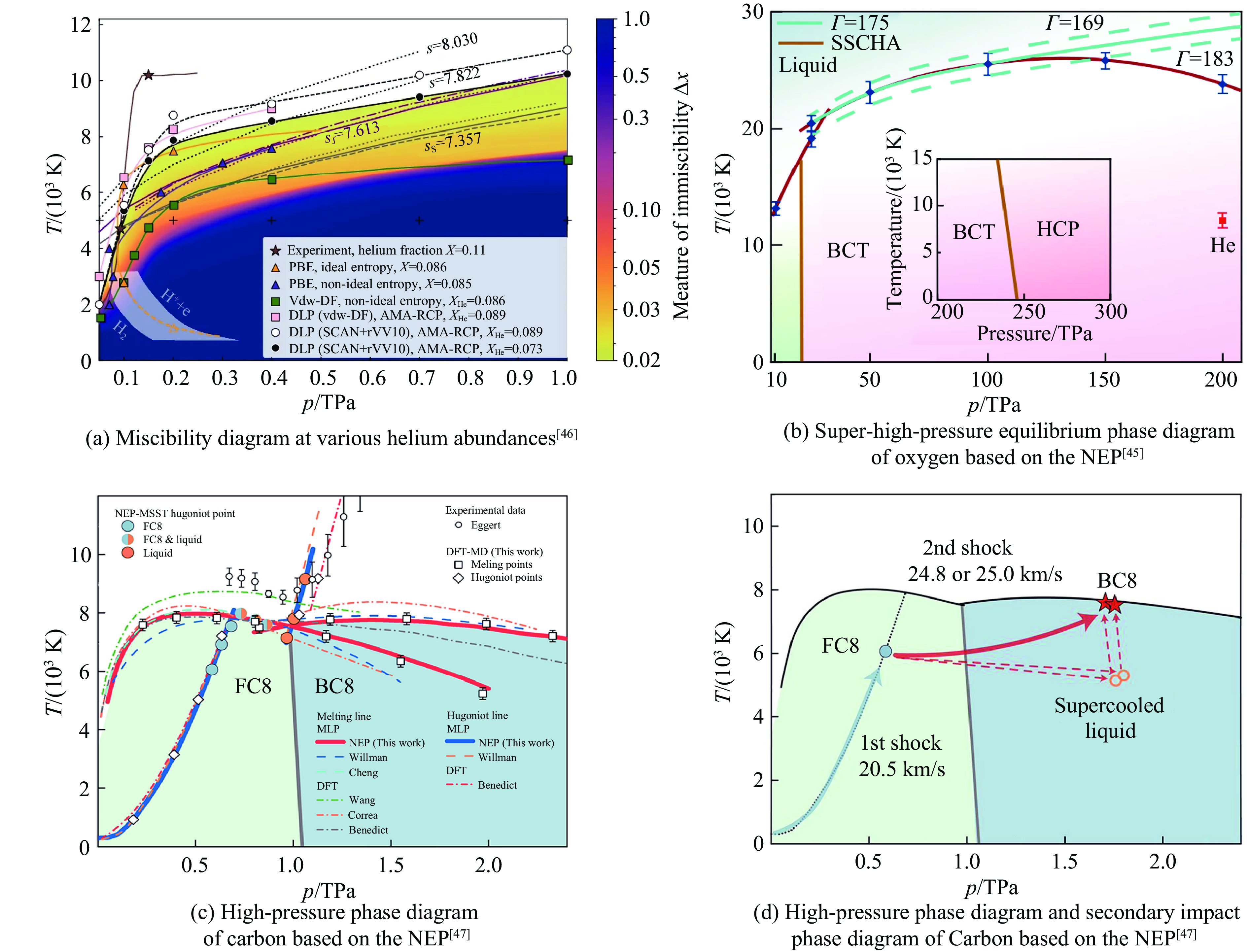
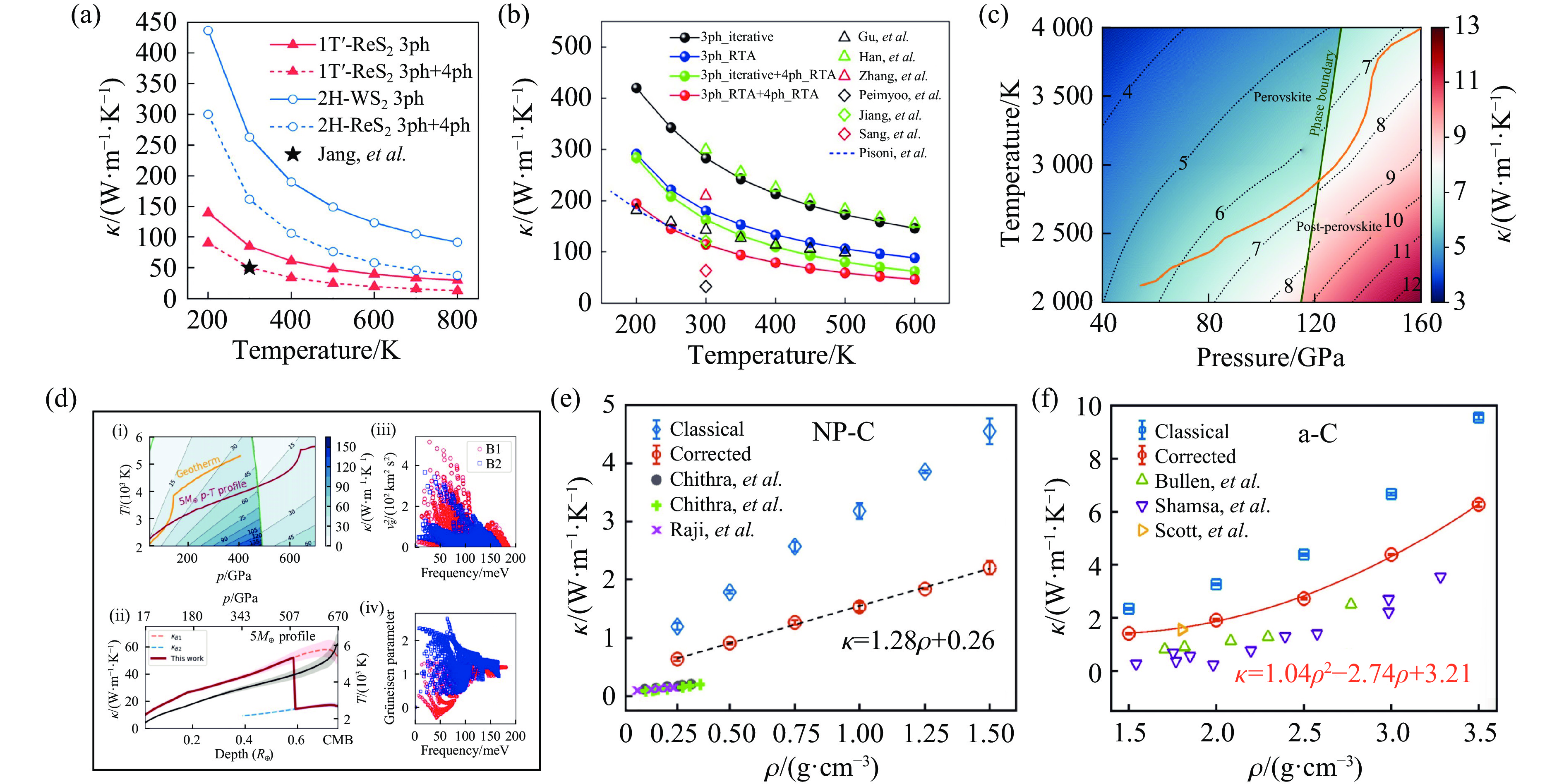
| Citation: | LI Jinlong, WANG Hao, GENG Huayun. A Review of Machine Learning Potentials in the Study of Materials Properties[J]. Chinese Journal of High Pressure Physics, 2026, 40(1): 010102. doi: 10.11858/gywlxb.20251172

|

| [1] |
CAR R, PARRINELLO M. Unified approach for molecular dynamics and density-functional theory [J]. Physical Review Letters, 1985, 55(22): 2471–2474. doi: 10.1103/PhysRevLett.55.2471
|
| [2] |
ALDER B J, WAINWRIGHT T E. Phase transition for a hard sphere system [J]. The Journal of Chemical Physics, 1957, 27(5): 1208–1209. doi: 10.1063/1.1743957
|
| [3] |
SHARIFI H, WICK C D. Developing interatomic potentials for complex concentrated alloys of Cu, Ti, Ni, Cr, Co, Al, Fe, and Mn [J]. Computational Materials Science, 2025, 248: 113595. doi: 10.1016/j.commatsci.2024.113595
|
| [4] |
MUELLER T, HERNANDEZ A, WANG C H. Machine learning for interatomic potential models [J]. The Journal of Chemical Physics, 2020, 152(5): 050902. doi: 10.1063/1.5126336
|
| [5] |
BEDOLLA E, PADIERNA L C, CASTAÑEDA-PRIEGO R. Machine learning for condensed matter physics [J]. Journal of Physics: Condensed Matter, 2020, 33(5): 053001. doi: 10.1088/1361-648X/abb895
|
| [6] |
CARLEO G, CIRAC I, CRANMER K, et al. Machine learning and the physical sciences [J]. Reviews of Modern Physics, 2019, 91(4): 045002. doi: 10.1103/RevModPhys.91.045002
|
| [7] |
BEHLER J, PARRINELLO M. Generalized neural-network representation of high-dimensional potential-energy surfaces [J]. Physical Review Letters, 2007, 98(14): 146401. doi: 10.1103/PhysRevLett.98.146401
|
| [8] |
SATORRAS V G, HOOGEBOOM E, WELLING M. E(n) equivariant graph neural networks [C]//Proceedings of the 38th International Conference on Machine Learning. PMLR, 2021: 9323–9332.
|
| [9] |
NOVIKOV I S, GUBAEV K, PODRYABINKIN E V, et al. The MLIP package: moment tensor potentials with MPI and active learning [J]. Machine Learning: Science and Technology, 2021, 2(2): 025002. doi: 10.1088/2632-2153/abc9fe
|
| [10] |
ZHANG Y Z, WANG H D, CHEN W J, et al. DP-GEN: a concurrent learning platform for the generation of reliable deep learning based potential energy models [J]. Computer Physics Communications, 2020, 253: 107206. doi: 10.1016/j.cpc.2020.107206
|
| [11] |
STENCZEL T K, EL-MACHACHI Z, LIEPUONIUTE G, et al. Machine-learned acceleration for molecular dynamics in CASTEP [J]. The Journal of Chemical Physics, 2023, 159(4): 044803. doi: 10.1063/5.0155621
|
| [12] |
JINNOUCHI R, KARSAI F, KRESSE G. On-the-fly machine learning force field generation: application to melting points [J]. Physical Review B, 2019, 100(1): 014105. doi: 10.1103/PhysRevB.100.014105
|
| [13] |
SHAPEEV A V. Moment tensor potentials: a class of systematically improvable interatomic potentials [J]. Multiscale Modeling & Simulation, 2016, 14(3): 1153–1173. doi: 10.1137/15M1054183
|
| [14] |
GUBAEV K, PODRYABINKIN E V, SHAPEEV A V. Machine learning of molecular properties: locality and active learning [J]. The Journal of Chemical Physics, 2018, 148(24): 241727. doi: 10.1063/1.5005095
|
| [15] |
WEN T Q, ZHANG L F, WANG H, et al. Deep potentials for materials science [J]. Materials Futures, 2022, 1(2): 022601. doi: 10.1088/2752-5724/ac681d
|
| [16] |
BARTÓK A P, KONDOR R, CSÁNYI G. On representing chemical environments [J]. Physical Review B, 2013, 87(18): 184115. doi: 10.1103/PhysRevB.87.184115
|
| [17] |
BEHLER J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials [J]. The Journal of Chemical Physics, 2011, 134(7): 074106. doi: 10.1063/1.3553717
|
| [18] |
BARTÓK A P, PAYNE M C, KONDOR R, et al. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons [J]. Physical Review Letters, 2010, 104(13): 136403. doi: 10.1103/PhysRevLett.104.136403
|
| [19] |
THOMPSON A P, SWILER L P, TROTT C R, et al. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials [J]. Journal of Computational Physics, 2015, 285: 316–330. doi: 10.1016/j.jcp.2014.12.018
|
| [20] |
ZHANG L F, HAN J Q, WANG H, et al. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics [J]. Physical Review Letters, 2018, 120(14): 143001. doi: 10.1103/PhysRevLett.120.143001
|
| [21] |
WANG H, ZHANG L F, HAN J Q, et al. DeePMD-kit: a deep learning package for many-body potential energy representation and molecular dynamics [J]. Computer Physics Communications, 2018, 228: 178–184. doi: 10.1016/j.cpc.2018.03.016
|
| [22] |
FAN Z Y, ZENG Z Z, ZHANG C Z, et al. Neuroevolution machine learning potentials: combining high accuracy and low cost in atomistic simulations and application to heat transport [J]. Physical Review B, 2021, 104(10): 104309. doi: 10.1103/PhysRevB.104.104309
|
| [23] |
HAN J, CEN J C, WU L M, et al. A survey of geometric graph neural networks: data structures, models and applications [J]. Frontiers of Computer Science, 2025, 19(11): 1911375. doi: 10.1007/s11704-025-41426-w
|
| [24] |
BATZNER S, MUSAELIAN A, SUN L X, et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials [J]. Nature Communications, 2022, 13(1): 2453. doi: 10.1038/s41467-022-29939-5
|
| [25] |
SCHÜTT K T, UNKE O T, GASTEGGER M. Equivariant message passing for the prediction of tensorial properties and molecular spectra [C]//Proceedings of the 38th International Conference on Machine Learning. PMLR, 2021: 9377–9388.
|
| [26] |
KLAWOHN S, DARBY J P, KERMODE J R, et al. Gaussian approximation potentials: theory, software implementation and application examples [J]. The Journal of Chemical Physics, 2023, 159(17): 174108. doi: 10.1063/5.0160898
|
| [27] |
DERINGER V L, BARTÓK A P, BERNSTEIN N, et al. Gaussian process regression for materials and molecules [J]. Chemical Reviews, 2021, 121(16): 10073–10141. doi: 10.1021/acs.chemrev.1c00022
|
| [28] |
CARO M A. Optimizing many-body atomic descriptors for enhanced computational performance of machine learning based interatomic potentials [J]. Physical Review B, 2019, 100(2): 024112. doi: 10.1103/PhysRevB.100.024112
|
| [29] |
YING P H, QIAN C, ZHAO R, et al. Advances in modeling complex materials: the rise of neuroevolution potentials [J]. Chemical Physics Reviews, 2025, 6(1): 011310. doi: 10.1063/5.0259061
|
| [30] |
GILMER J, SCHOENHOLZ S S, RILEY P F, et al. Neural message passing for quantum chemistry [C]//Proceedings of the 34th International Conference on Machine Learning. Sydney: JMLR. org, 2017: 1263–1272.
|
| [31] |
SCHÜTT K T, SAUCEDA H E, KINDERMANS P J, et al. SchNet-a deep learning architecture for molecules and materials [J]. The Journal of Chemical Physics, 2018, 148(24): 241722. doi: 10.1063/1.5019779
|
| [32] |
BATATIA I, KOVÁCS D P, SIMM G N C, et al. MACE: higher order equivariant message passing neural networks for fast and accurate force fields [C]//Proceedings of the 36th International Conference on Neural Information Processing Systems. New Orleans: Curran Associates Inc. , 2022: 830.
|
| [33] |
YANG Z D, WANG X, LI Y F, et al. Efficient equivariant model for machine learning interatomic potentials [J]. NPJ Computational Materials, 2025, 11(1): 49. doi: 10.1038/s41524-025-01535-3
|
| [34] |
ZUO Y X, CHEN C, LI X G, et al. Performance and cost assessment of machine learning interatomic potentials [J]. The Journal of Physical Chemistry A, 2020, 124(4): 731–745. doi: 10.1021/acs.jpca.9b08723
|
| [35] |
FAN Z Y, WANG Y Z, YING P H, et al. GPUMD: a package for constructing accurate machine-learned potentials and performing highly efficient atomistic simulations [J]. The Journal of Chemical Physics, 2022, 157(11): 114801. doi: 10.1063/5.0106617
|
| [36] |
WILLMAN J T, GONZALEZ J M, NGUYEN-CONG K, et al. Accuracy, transferability, and computational efficiency of interatomic potentials for simulations of carbon under extreme conditions [J]. The Journal of Chemical Physics, 2024, 161(8): 084709. doi: 10.1063/5.0218705
|
| [37] |
MO P H, ZHANG Y J, ZHAO Z Y, et al. High-speed and low-power molecular dynamics processing unit (MDPU) with ab initio accuracy [J]. NPJ Computational Materials, 2024, 10(1): 253. doi: 10.1038/s41524-024-01422-3
|
| [38] |
MENG Z J, ZONGO K, TORRES E, et al. A Kokkos-accelerated moment tensor potential implementation for LAMMPS [EB/OL]. arXiv: 2510.00193. (2025-09-30)[2025-08-26]. https://arxiv.org/abs/2510.00193.
|
| [39] |
HUANG J, RATHOD V, SUN C, et al. Speed/accuracy trade-offs for modern convolutional object detectors [C]//Proceedings of the 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR). Honolulu: IEEE, 2017: 3296–3297.
|
| [40] |
WANG J T, LIU P T, ZHU H Y, et al. Efficient moment tensor machine-learning interatomic potential for accurate description of defects in Ni-Al alloys [J]. Physical Review Materials, 2025, 9(5): 053805. doi: 10.1103/PhysRevMaterials.9.053805
|
| [41] |
WILLMAN J T, NGUYEN-CONG K, WILLIAMS A S, et al. Machine learning interatomic potential for simulations of carbon at extreme conditions [J]. Physical Review B, 2022, 106(18): L180101. doi: 10.1103/PhysRevB.106.L180101
|
| [42] |
KRUGLOV I A, YANILKIN A, OGANOV A R, et al. Phase diagram of uranium from ab initio calculations and machine learning [J]. Physical Review B, 2019, 100(17): 174104. doi: 10.1103/PhysRevB.100.174104
|
| [43] |
CHIRKOV P V, ELTSOV G S, KICHIGIN R M, et al. Machine learning enhanced quantum molecular dynamics assessment of the phase diagram of uranium [J]. Physical Review B, 2025, 112(2): 024104. doi: 10.1103/8w5w-f3kd
|
| [44] |
ZHANG L F, WANG H, CAR R, et al. Phase diagram of a deep potential water model [J]. Physical Review Letters, 2021, 126(23): 236001. doi: 10.1103/PhysRevLett.126.236001
|
| [45] |
WANG Y L, SHI J Y, LIANG Z X, et al. Machine learning simulations reveal oxygen’s phase diagram and thermal properties at conditions relevant to white dwarfs [J]. Nature Communications, 2025, 16(1): 5504. doi: 10.1038/s41467-025-61390-0
|
| [46] |
CHANG X J, CHEN B, ZENG Q Y, et al. Theoretical evidence of H-He demixing under Jupiter and Saturn conditions [J]. Nature Communications, 2024, 15(1): 8543. doi: 10.1038/s41467-024-52868-4
|
| [47] |
SHI J Y, LIANG Z X, WANG J J, et al. Double-shock compression pathways from diamond to BC8 carbon [J]. Physical Review Letters, 2023, 131(14): 146101. doi: 10.1103/PhysRevLett.131.146101
|
| [48] |
ZONG H X, LUO Y F, DING X D, et al. hcp→ω phase transition mechanisms in shocked zirconium: a machine learning based atomic simulation study [J]. Acta Materialia, 2019, 162: 126–135. doi: 10.1016/j.actamat.2018.09.067
|
| [49] |
ZONG H X, PILANIA G, DING X D, et al. Developing an interatomic potential for martensitic phase transformations in zirconium by machine learning [J]. NPJ Computational Materials, 2018, 4(1): 48. doi: 10.1038/s41524-018-0103-x
|
| [50] |
ZHU S C, CHEN G W, YUAN X H, et al. Key for hexagonal diamond formation: theoretical and experimental study [J]. Journal of the American Chemical Society, 2025, 147(2): 2158–2167. doi: 10.1021/jacs.4c16312
|
| [51] |
CHEN B, ZENG Q Y, WANG H, et al. Atomistic mechanism of phase transition in shock compressed gold revealed by deep potential [EB/OL]. arXiv: 2006.13136. (2021-07-19)[2025-08-26]. https://arxiv.org/abs/2006.13136.
|
| [52] |
ZHU L F, KÖRMANN F, CHEN Q, et al. Accelerating ab initio melting property calculations with machine learning: application to the high entropy alloy TaVCrW [J]. NPJ Computational Materials, 2024, 10(1): 274. doi: 10.1038/s41524-024-01464-7
|
| [53] |
NITOL M S, MISHRA A, XU S Z, et al. Moment tensor potential and its application in the Ti-Al-V multicomponent system [J]. Physical Review Materials, 2025, 9(6): 063601. doi: 10.1103/PhysRevMaterials.9.063601
|
| [54] |
ROSENBROCK C W, GUBAEV K, SHAPEEV A V, et al. Machine-learned interatomic potentials for alloys and alloy phase diagrams [J]. NPJ Computational Materials, 2021, 7(1): 24. doi: 10.1038/s41524-020-00477-2
|
| [55] |
JUNG J H, FORSLUND A, SRINIVASAN P, et al. Dynamically stabilized phases with full ab initio accuracy: thermodynamics of Ti, Zr, Hf with a focus on the hcp-bcc transition [J]. Physical Review B, 2023, 108(18): 184107. doi: 10.1103/PhysRevB.108.184107
|
| [56] |
HUANG Y F, YAO S L, CHEN H J, et al. An atomistic study of γ→γ0→α" martensitic transformation in uranium-6 wt% niobium [J]. Journal of Nuclear Materials, 2025, 606: 155617. doi: 10.1016/j.jnucmat.2025.155617
|
| [57] |
ZHAO J L, BYGGMÄSTAR J, ZHANG Z F, et al. Phase transition of two-dimensional ferroelectric and paraelectric Ga2O3 monolayers: a density functional theory and machine learning study [J]. Physical Review B, 2021, 104(5): 054107. doi: 10.1103/PhysRevB.104.054107
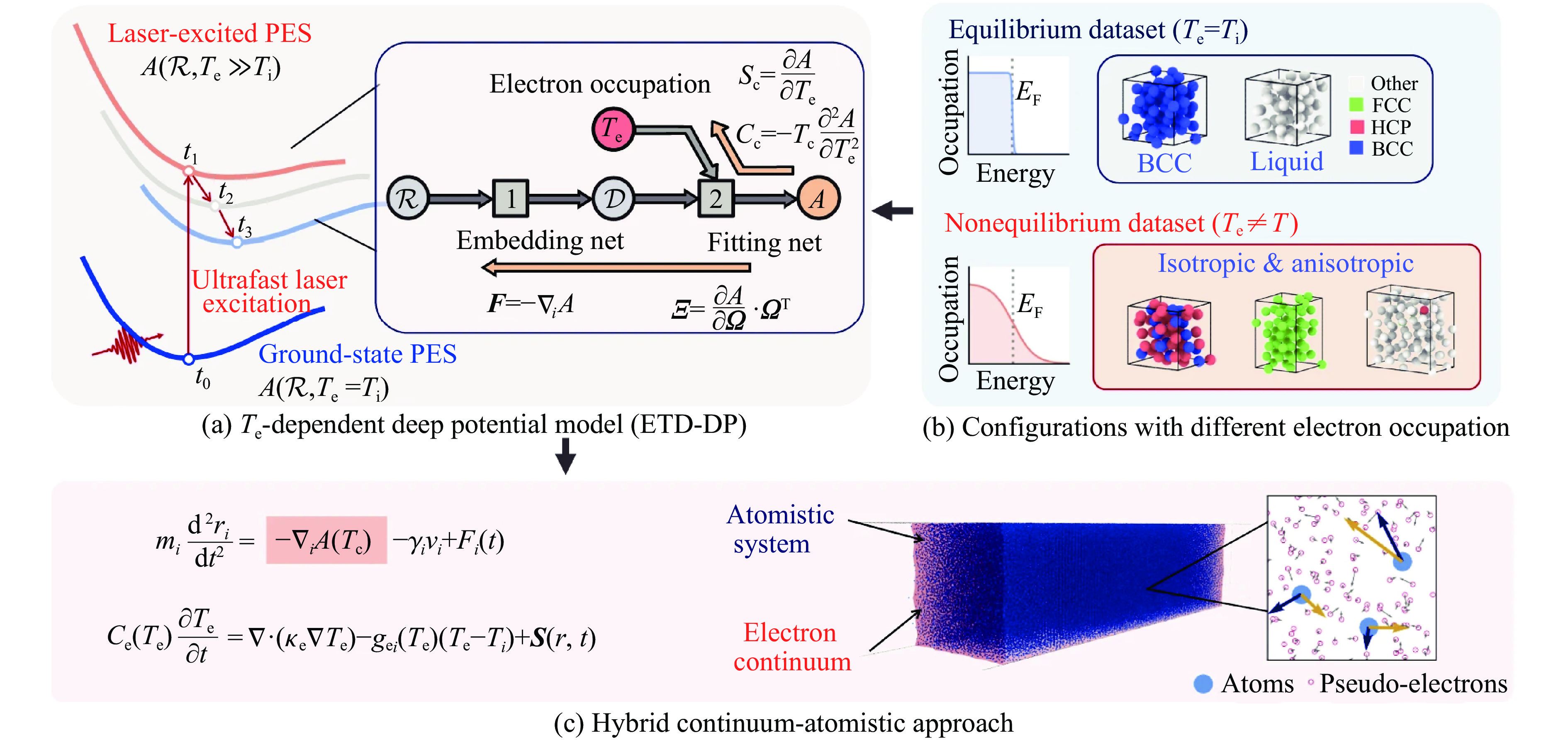
|
| [58] |
ERHARD L C, ROHRER J, ALBE K, et al. A machine-learned interatomic potential for silica and its relation to empirical models [J]. NPJ Computational Materials, 2022, 8(1): 90. doi: 10.1038/s41524-022-00768-w/
|
| [59] |
ZHAO J L, BYGGMÄSTAR J, HE H, et al. Complex Ga2O3 polymorphs explored by accurate and general-purpose machine-learning interatomic potentials [J]. NPJ Computational Materials, 2023, 9(1): 159. doi: 10.1038/s41524-023-01117-1
|
| [60] |
LI X G, HU C Z, CHEN C, et al. Quantum-accurate spectral neighbor analysis potential models for Ni-Mo binary alloys and fcc metals [J]. Physical Review B, 2018, 98(9): 094104. doi: 10.1103/PhysRevB.98.094104
|
| [61] |
RICHARD P, CASTELLANO A, BÉJAUD R, et al. Ab initio phase diagram of gold in extreme conditions [J]. Physical Review Letters, 2023, 131(20): 206101. doi: 10.1103/PhysRevLett.131.206101
|
| [62] |
BIDEAULT M, CREUZE J, ASAHI R, et al. Polyvalent machine-learned potential for cobalt: from bulk to nanoparticles [J]. Physical Review Materials, 2024, 8(12): 123803. doi: 10.1103/PhysRevMaterials.8.123803
|
| [63] |
LIU P T, VERDI C, KARSAI F, et al. Phase transitions of zirconia: machine-learned force fields beyond density functional theory [J]. Physical Review B, 2022, 105(6): L060102. doi: 10.1103/PhysRevB.105.L060102
|
| [64] |
LIU P T, VERDI C, KARSAI F, et al. α−β phase transition of zirconium predicted by on-the-fly machine-learned force field [J]. Physical Review Materials, 2021, 5(5): 053804. doi: 10.1103/PhysRevMaterials.5.053804
|
| [65] |
NIU H W, YANG Y B, JENSEN S, et al. Stable solid molecular hydrogen above 900 K from a machine-learned potential trained with diffusion quantum Monte Carlo [J]. Physical Review Letters, 2023, 130(7): 076102. doi: 10.1103/PhysRevLett.130.076102
|
| [66] |
CHEN T, YUAN F B, LIU J C, et al. Modeling the high-pressure solid and liquid phases of tin from deep potentials with ab initio accuracy [J]. Physical Review Materials, 2023, 7(5): 053603. doi: 10.1103/PhysRevMaterials.7.053603
|
| [67] |
DENG J, NIU H Y, HU J W, et al. Melting of MgSiO3 determined by machine learning potentials [J]. Physical Review B, 2023, 107(6): 064103. doi: 10.1103/PhysRevB.107.064103
|
| [68] |
GUO L F, JIN T, SHAN S, et al. Structural transformations in single-crystalline AgPd nanoalloys from multiscale deep potential molecular dynamics [J]. The Journal of Chemical Physics, 2023, 159(2): 024702. doi: 10.1063/5.0158918
|
| [69] |
WISESA P, ANDOLINA C M, SAIDI W A. Machine-learning accelerated first-principles accurate modeling of the solid-liquid phase transition in MgO under mantle conditions [J]. The Journal of Physical Chemistry Letters, 2023, 14(39): 8741–8748. doi: 10.1021/acs.jpclett.3c02424
|
| [70] |
HE R, WU H Y, ZHANG L F, et al. Structural phase transitions in SrTiO3 from deep potential molecular dynamics [J]. Physical Review B, 2022, 105(6): 064104. doi: 10.1103/PhysRevB.105.064104
|
| [71] |
WEN T Q, WANG R, ZHU L Y, et al. Specialising neural network potentials for accurate properties and application to the mechanical response of titanium [J]. NPJ Computational Materials, 2021, 7(1): 206. doi: 10.1038/s41524-021-00661-y
|
| [72] |
NIU H Y, BONATI L, PIAGGI P M, et al. Ab initio phase diagram and nucleation of gallium [J]. Nature Communications, 2020, 11(1): 2654. doi: 10.1038/s41467-020-16372-9
|
| [73] |
FENG X K, PAN S N, KATAGIRI K, et al. Nanosecond structural evolution in shocked coesite [J]. Science Advances, 2025, 11(17): eads3139. doi: 10.1126/sciadv.ads3139
|
| [74] |
KAYASTHA P, FRANSSON E, ERHART P, et al. Octahedral tilt-driven phase transitions in BaZrS3 chalcogenide perovskite [J]. The Journal of Physical Chemistry Letters, 2025, 16(8): 2064–2071. doi: 10.1021/acs.jpclett.4c03517
|
| [75] |
PAN S N, SHI J Y, LIANG Z X, et al. Shock compression pathways to pyrite silica from machine learning simulations [J]. Physical Review B, 2024, 110(22): 224101. doi: 10.1103/PhysRevB.110.224101
|
| [76] |
FRANSSON E, RAHM J M, WIKTOR J, et al. Revealing the free energy landscape of halide perovskites: metastability and transition characters in CsPbBr3 and MAPbI3 [J]. Chemistry of Materials, 2023, 35(19): 8229–8238. doi: 10.1021/acs.chemmater.3c01740
|
| [77] |
PAN S N, HUANG T H, VAZAN A, et al. Magnesium oxide-water compounds at megabar pressure and implications on planetary interiors [J]. Nature Communications, 2023, 14(1): 1165. doi: 10.1038/s41467-023-36802-8
|
| [78] |
ISTAS M, JENSEN S, YANG Y B, et al. Liquid-liquid phase transition of hydrogen and its critical point: analysis from ab initio simulation and a machine-learned potential [J]. Physical Review E, 2025, 111(4): 045307. doi: 10.1103/PhysRevE.111.045307
|
| [79] |
LY K K, CEPERLEY D M. Melting curves of atomic hydrogen and deuterium calculated using path-integral Monte Carlo [J]. Physical Review B, 2025, 111(10): 104102. doi: 10.1103/PhysRevB.111.104102
|
| [80] |
YANG X, CHEN Y S, ZHENG Y H, et al. Understanding the importance of four-phonon scattering in low-symmetry monolayer 1T′-ReS2 using machine learning potential [J]. Applied Physics Letters, 2024, 124(7): 072204. doi: 10.1063/5.0190570
|
| [81] |
ZHANG G W, DONG S L, YANG C, et al. Revisiting four-phonon scattering in WS2 monolayer with machine learning potential [J]. Applied Physics Letters, 2023, 123(5): 052205. doi: 10.1063/5.0159517
|
| [82] |
YANG F H, ZENG Q Y, CHEN B, et al. Lattice thermal conductivity of MgSiO3 perovskite and post-perovskite under lower mantle conditions calculated by deep potential molecular dynamics [J]. Chinese Physics Letters, 2022, 39(11): 116301. doi: 10.1088/0256-307X/39/11/116301
|
| [83] |
QIU R, ZENG Q Y, HAN J S, et al. Coupled temperature-density dependence of lattice thermal conductivity of MgO at extreme conditions [J]. Physical Review B, 2025, 111(6): 064103. doi: 10.1103/PhysRevB.111.064103
|
| [84] |
WANG Y Z, FAN Z Y, QIAN P, et al. Density dependence of thermal conductivity in nanoporous and amorphous carbon with machine-learned molecular dynamics [J]. Physical Review B, 2025, 111(9): 094205. doi: 10.1103/PhysRevB.111.094205
|
| [85] |
OUYANG Y L, YU C Q, HE J, et al. Accurate description of high-order phonon anharmonicity and lattice thermal conductivity from molecular dynamics simulations with machine learning potential [J]. Physical Review B, 2022, 105(11): 115202. doi: 10.1103/PhysRevB.105.115202
|
| [86] |
KOROTAEV P, NOVOSELOV I, YANILKIN A, et al. Accessing thermal conductivity of complex compounds by machine learning interatomic potentials [J]. Physical Review B, 2019, 100(14): 144308. doi: 10.1103/PhysRevB.100.144308
|
| [87] |
WANG Q, ZENG Z Z, CHEN Y. Revisiting phonon transport in perovskite SrTiO3: anharmonic phonon renormalization and four-phonon scattering [J]. Physical Review B, 2021, 104(23): 235205. doi: 10.1103/PhysRevB.104.235205
|
| [88] |
MORTAZAVI B, PODRYABINKIN E V, NOVIKOV I S, et al. Accelerating first-principles estimation of thermal conductivity by machine-learning interatomic potentials: a MTP/ShengBTE solution [J]. Computer Physics Communications, 2021, 258: 107583. doi: 10.1016/j.cpc.2020.107583
|
| [89] |
WANG D Y, ZHAO K, HONG T, et al. Intrinsically low lattice thermal conductivity and multivalley band structure induced promising high thermoelectric performance in Pb3Bi2S6 [J]. Materials Today Physics, 2025, 51: 101654. doi: 10.1016/j.mtphys.2025.101654
|
| [90] |
LIU H, QIAN X, BAO H, et al. High-temperature phonon transport properties of SnSe from machine-learning interatomic potential [J]. Journal of Physics: Condensed Matter, 2021, 33(40): 405401. doi: 10.1088/1361-648X/ac13fd
|
| [91] |
LI Y H, LI X, WEI B, et al. Phonon coherence in bismuth-halide perovskite Cs3Bi2Br9 with ultralow thermal conductivity [J]. Advanced Functional Materials, 2024, 34(52): 2411152. doi: 10.1002/adfm.202411152
|
| [92] |
ZHANG Y X, SHEN C, LONG T, et al. Thermal conductivity of h-BN monolayers using machine learning interatomic potential [J]. Journal of Physics: Condensed Matter, 2021, 33(10): 105903. doi: 10.1088/1361-648X/abcf61
|
| [93] |
REN J L, ZHANG G H, WANG X M, et al. Research on machine learning interatomic potentials for titanium oxide ceramic materials [J]. Physica B: Condensed Matter, 2025, 711: 417281. doi: 10.1016/j.physb.2025.417281
|
| [94] |
TANG J L, LI G T, WANG Q, et al. Effect of four-phonon scattering on anisotropic thermal transport in bulk hexagonal boron nitride by machine learning interatomic potential [J]. International Journal of Heat and Mass Transfer, 2023, 207: 124011. doi: 10.1016/j.ijheatmasstransfer.2023.124011
|
| [95] |
TANG J L, LI G T, WANG Q, et al. Competition between phonon-vacancy and four-phonon scattering in cubic boron arsenide by machine learning interatomic potential [J]. Physical Review Materials, 2023, 7(4): 044601. doi: 10.1103/PhysRevMaterials.7.044601
|
| [96] |
BABAEI H, GUO R Q, HASHEMI A, et al. Machine-learning-based interatomic potential for phonon transport in perfect crystalline Si and crystalline Si with vacancies [J]. Physical Review Materials, 2019, 3(7): 074603. doi: 10.1103/PhysRevMaterials.3.074603
|
| [97] |
GU X K, ZHAO C Y. Thermal conductivity of single-layer MoS2(1−x)Se2x alloys from molecular dynamics simulations with a machine-learning-based interatomic potential [J]. Computational Materials Science, 2019, 165: 74–81. doi: 10.1016/j.commatsci.2019.04.025
|
| [98] |
ZHANG Y H, LUNGHI A, SANVITO S. Pushing the limits of atomistic simulations towards ultra-high temperature: a machine-learning force field for ZrB2 [J]. Acta Materialia, 2020, 186: 467–474. doi: 10.1016/j.actamat.2019.12.060
|
| [99] |
LAHNSTEINER J, RANG M, BOKDAM M. Tuning einstein oscillator frequencies of cation rattlers: a molecular dynamics study of the lattice thermal conductivity of CsPbBr3 [J]. The Journal of Physical Chemistry C. Nanomaterials and Interfaces, 2024, 128(3): 1341–1349. doi: 10.1021/acs.jpcc.3c06590
|
| [100] |
CUI C F, ZHANG Y W, OUYANG T, et al. On-the-fly machine learning potential accelerated accurate prediction of lattice thermal conductivity of metastable silicon crystals [J]. Physical Review Materials, 2023, 7(3): 033803. doi: 10.1103/PhysRevMaterials.7.033803
|
| [101] |
QIU R, ZENG Q Y, WANG H, et al. Anomalous thermal transport across the superionic transition in ice [J]. Chinese Physics Letters, 2023, 40(11): 116301. doi: 10.1088/0256-307X/40/11/116301
|
| [102] |
BHATT N, KARNA P, THAKUR S, et al. Pressure-driven enhancement of phonon contribution to the thermal conductivity of Iridium [J]. International Journal of Heat and Mass Transfer, 2024, 229: 125673. doi: 10.1016/j.ijheatmasstransfer.2024.125673
|
| [103] |
CHE J W, HUANG W J, REN G L, et al. Dual-channel phonon transport leads to low thermal conductivity in pyrochlore La2Hf2O7 [J]. Ceramics International, 2024, 50(13): 22865–22873. doi: 10.1016/j.ceramint.2024.04.011
|
| [104] |
MALGOPE S, GUPTA M K, BAG S, et al. Untangling high-temperature thermal expansion and lattice thermal conductivity behavior of vanadium using machine-learned molecular dynamics [J]. Physical Review B, 2024, 110(5): 054301. doi: 10.1103/PhysRevB.110.054301
|
| [105] |
PENG Y H, DENG J. Thermal conductivity of MgSiO3-H2O system determined by machine learning potentials [J]. Geophysical Research Letters, 2024, 51(5): e2023GL107245. doi: 10.1029/2023GL107245
|
| [106] |
ZHANG P, JIN D, QIN M, et al. Effects of four-phonon interaction and vacancy defects on the thermal conductivity of the low-temperature phase of SnSe [J]. Physical Review Applied, 2024, 21(2): 024043. doi: 10.1103/PhysRevApplied.21.024043
|
| [107] |
WANG D, WU Z Q, DENG X. Thermal conductivity of hydrous wadsleyite determined by non-equilibrium molecular dynamics based on machine learning [J]. Geophysical Research Letters, 2022, 49(22): e2022GL100337. doi: 10.1029/2022GL100337
|
| [108] |
MIRCHI P, ADESSI C, MERABIA S, et al. Lattice thermal conductivity and mechanical properties of the single-layer penta-NiN2 explored by a deep-learning interatomic potential [J]. Physical Chemistry Chemical Physics, 2024, 26(19): 14216–14227. doi: 10.1039/D4CP00997E
|
| [109] |
ZHANG P, ZHANG Z H, LIU Y, et al. Phonon thermal transport in Bi2Te3 from a deep-neural-network interatomic potential [J]. Physical Review Applied, 2022, 18(5): 054022. doi: 10.1103/PhysRevApplied.18.054022
|
| [110] |
HU S, GU X K. Approaching to low thermal conductivity limit in layered materials through full-spectrum phonon band engineering [J]. Materials Today Physics, 2025, 52: 101669. doi: 10.1016/j.mtphys.2025.101669
|
| [111] |
LI K, MA H. Decoding the thermal conductivity of ionic covalent organic frameworks: optical phonons as key determinants revealed by neuroevolution potential [J]. Materials Today Physics, 2025, 54: 101724. doi: 10.1016/j.mtphys.2025.101724
|
| [112] |
ZHOU X T, XU Y J, CHEN Y, et al. Mechanism on lattice thermal conductivity of carbon-vacancy and porous medium entropy ceramics [J]. Scripta Materialia, 2025, 259: 116568. doi: 10.1016/j.scriptamat.2025.116568
|
| [113] |
WANG Y, XIE L, YANG H B, et al. Strong orbital-lattice coupling induces glassy thermal conductivity in high-symmetry single crystal BaTiS3 [J]. Physical Review X, 2025, 15(1): 011066. doi: 10.1103/PhysRevX.15.011066
|
| [114] |
LIU Y W, FU Y M, GU F C, et al. Lattice-distortion-driven reduced lattice thermal conductivity in high-entropy ceramics [J]. Advanced Science, 2025, 12(19): 2501157. doi: 10.1002/advs.202501157
|
| [115] |
CAO C Y, CAO S, ZHU Y X, et al. Thermal transports of 2D phosphorous carbides by machine learning molecular dynamics simulations [J]. International Journal of Heat and Mass Transfer, 2024, 224: 125359. doi: 10.1016/j.ijheatmasstransfer.2024.125359
|
| [116] |
YANG J, ZHU X Y, MCGAUGHEY A J H, et al. Two-mode terms in Wigner transport equation elucidate anomalous thermal transport in amorphous silicon [J]. Physical Review B, 2025, 111(9): 094206. doi: 10.1103/PhysRevB.111.094206
|
| [117] |
FAN H Z, YING P H, FAN Z Y, et al. Anomalous strain-dependent thermal conductivity in the metal-organic framework HKUST-1 [J]. Physical Review B, 2024, 109(4): 045424. doi: 10.1103/PhysRevB.109.045424
|
| [118] |
HUANG X F, LI C Z, YUAN M H, et al. Unphysical grain size dependence of lattice thermal conductivity in Mg3(Sb, Bi)2: an atomistic view of concentration dependent segregation effects [J]. Materials Today Physics, 2024, 43: 101386. doi: 10.1016/j.mtphys.2024.101386
|
| [119] |
SO S, LEE J H. Unraveling interfacial thermal transport in β-Ga2O3/h-BN van der Waals heterostructures [J]. Materials Today Physics, 2024, 46: 101506. doi: 10.1016/j.mtphys.2024.101506
|
| [120] |
LI W, CARRETE J, KATCHO N A, et al. ShengBTE: a solver of the Boltzmann transport equation for phonons [J]. Computer Physics Communications, 2014, 185(6): 1747–1758. doi: 10.1016/j.cpc.2014.02.015
|
| [121] |
KUBO R. Statistical-mechanical theory of irreversible processes. Ⅰ. general theory and simple applications to magnetic and conduction problems [J]. Journal of the Physical Society of Japan, 1957, 12(6): 570–586. doi: 10.1143/JPSJ.12.570
|
| [122] |
LI Z, XIONG S Y, SIEVERS C, et al. Influence of thermostatting on nonequilibrium molecular dynamics simulations of heat conduction in solids [J]. The Journal of Chemical Physics, 2019, 151(23): 234105. doi: 10.1063/1.5132543
|
| [123] |
FAN Z Y, DONG H K, HARJU A, et al. Homogeneous nonequilibrium molecular dynamics method for heat transport and spectral decomposition with many-body potentials [J]. Physical Review B, 2019, 99(6): 064308. doi: 10.1103/PhysRevB.99.064308
|
| [124] |
DONG H K, SHI Y B, YING P H, et al. Molecular dynamics simulations of heat transport using machine-learned potentials: a mini-review and tutorial on GPUMD with neuroevolution potentials [J]. Journal of Applied Physics, 2024, 135(16): 161101. doi: 10.1063/5.0200833
|
| [125] |
WANG H, PAN X L, WANG Y F, et al. Lattice dynamics and elastic properties of α-U at high-temperature and high-pressure by machine learning potential simulations [J]. Journal of Nuclear Materials, 2022, 572: 154029. doi: 10.1016/j.jnucmat.2022.154029
|
| [126] |
ZHANG C, YANG J Y, SUN T, et al. Strong precursor softening in cubic CaSiO3 perovskite [J]. Proceedings of the National Academy of Sciences of the United States of America, 2025, 122(5): e2410910122. doi: 10.1073/pnas.2410910122
|
| [127] |
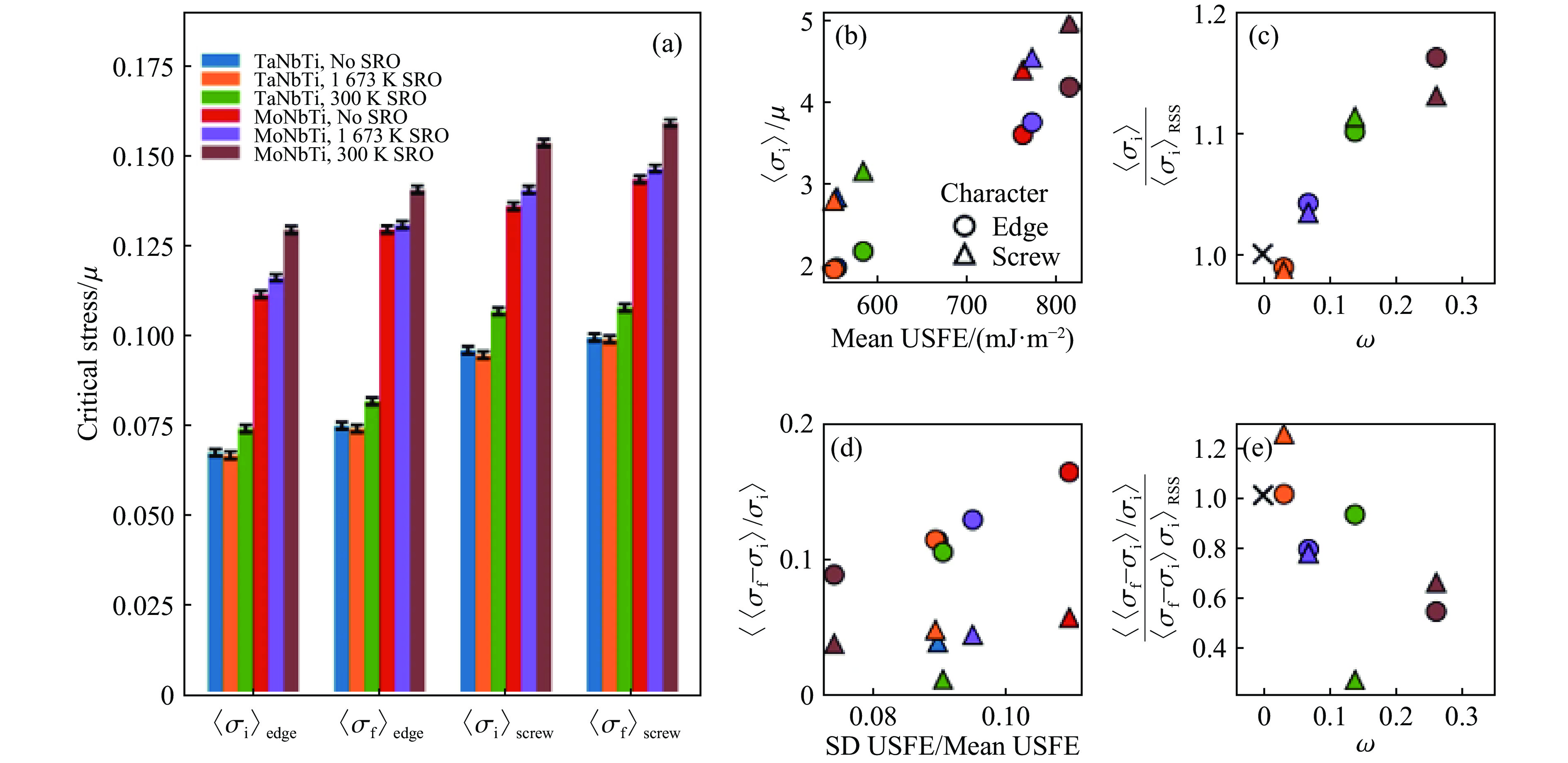
ZHENG H, FEY L T W, LI X G, et al. Multi-scale investigation of short-range order and dislocation glide in MoNbTi and TaNbTi multi-principal element alloys [J]. NPJ Computational Materials, 2023, 9(1): 89. doi: 10.1038/s41524-023-01046-z
|
| [128] |
WU L P, LI T. A machine learning interatomic potential for high entropy alloys [J]. Journal of the Mechanics and Physics of Solids, 2024, 187: 105639. doi: 10.1016/j.jmps.2024.105639
|
| [129] |
SHAPEEV A V, PODRYABINKIN E V, GUBAEV K, et al. Elinvar effect in β-Ti simulated by on-the-fly trained moment tensor potential [J]. New Journal of Physics, 2020, 22(11): 113005. doi: 10.1088/1367-2630/abc392
|
| [130] |
ZOTOV N, GUBAEV K, WÖRNER J, et al. Moment tensor potential for static and dynamic investigations of screw dislocations in bcc Nb [J]. Modelling and Simulation in Materials Science and Engineering, 2024, 32(3): 035032. doi: 10.1088/1361-651X/ad2d68
|
| [131] |
GUBAEV K, IKEDA Y, TASNÁDI F, et al. Finite-temperature interplay of structural stability, chemical complexity, and elastic properties of bcc multicomponent alloys from ab initio trained machine-learning potentials [J]. Physical Review Materials, 2021, 5(7): 073801. doi: 10.1103/PhysRevMaterials.5.073801
|
| [132] |
JALOLOV F N, PODRYABINKIN E V, OGANOV A R, et al. Mechanical properties of single and polycrystalline solids from machine learning [J]. Advanced Theory and Simulations, 2024, 7(5): 2301171. doi: 10.1002/adts.202301171
|
| [133] |
YANG G L, PENG H X, HUANG J, et al. Atomic perspective on plasticity mechanism of ionic-covalent silver sulfide [J]. International Journal of Mechanical Sciences, 2025, 300: 110471. doi: 10.1016/j.ijmecsci.2025.110471
|
| [134] |
CHOYAL V, MISHRA S, LUHADIYA N, et al. Development and evaluation of machine-learned interatomic potentials for carbon nanotubes for molecular dynamics simulations [J]. Carbon Letters, 2025, 35(3): 1311–1326. doi: 10.1007/s42823-025-00867-w
|
| [135] |
CHOYAL V, PATIL M, LUHADIYA N, et al. Machine-learned interatomic potentials for accurate analysis of the mechanical properties of boron nitride sheets [J]. Journal of Physics: Materials, 2025, 8(1): 015003. doi: 10.1088/2515-7639/ad9635
|
| [136] |
MUKHAMEDOV B, TASNÁDI F, ABRIKOSOV I A. Machine learning interatomic potential for the low-modulus Ti-Nb-Zr alloys in the vicinity of dynamical instability [J]. Materials & Design, 2025, 253: 113865. doi: 10.1016/j.matdes.2025.113865
|
| [137] |
OURDANI D, CASTELLANO A, VYTHELINGUM A K, et al. Experimental determination of the temperature- and phase-dependent elastic constants of FeRh [J]. Physical Review B, 2024, 110(1): 014427. doi: 10.1103/PhysRevB.110.014427
|
| [138] |
KOSKENNIEMI M, BYGGMÄSTAR J, NORDLUND K, et al. Efficient atomistic simulations of radiation damage in W and W-Mo using machine-learning potentials [J]. Journal of Nuclear Materials, 2023, 577: 154325. doi: 10.1016/j.jnucmat.2023.154325
|
| [139] |
WANG X W, XU S Z, JIAN W R, et al. Generalized stacking fault energies and Peierls stresses in refractory body-centered cubic metals from machine learning-based interatomic potentials [J]. Computational Materials Science, 2021, 192: 110364. doi: 10.1016/j.commatsci.2021.110364
|
| [140] |
DUBOIS E T, TRANCHIDA J, BOUCHET J, et al. Atomistic simulations of nuclear fuel UO2 with machine learning interatomic potentials [J]. Physical Review Materials, 2024, 8(2): 025402. doi: 10.1103/PhysRevMaterials.8.025402
|
| [141] |
HOSSAIN R, KIMIZUKA H, OGATA S. Asymmetry in core structure and mobility of basal dislocations in a Ti3SiC2 MAX phase: an atomistic study with machine-learned force fields [J]. Physical Review Materials, 2023, 7(5): 053608. doi: 10.1103/PhysRevMaterials.7.053608
|
| [142] |
YANG W, YE J W, BI P, et al. Mechanical properties of Mo-Re alloy based on first-principles and machine learning potential function [J]. Materials Today Communications, 2024, 38: 107796. doi: 10.1016/j.mtcomm.2023.107796
|
| [143] |
WAN S J, TONG R T, HAN B, et al. Machine learning interatomic potential for friction study in silicon and molybdenum disulfide [J]. Computational Materials Science, 2025, 248: 113608. doi: 10.1016/j.commatsci.2024.113608
|
| [144] |
WANG X Y, WANG Y N, ZHANG L F, et al. A tungsten deep neural-network potential for simulating mechanical property degradation under fusion service environment [J]. Nuclear Fusion, 2022, 62(12): 126013. doi: 10.1088/1741-4326/ac888b
|
| [145] |
WAN T Q, LUO C X, SUN Y, et al. Thermoelastic properties of bridgmanite using deep-potential molecular dynamics [J]. Physical Review B, 2024, 109(9): 094101. doi: 10.1103/PhysRevB.109.094101
|
| [146] |
FAN D, NASKAR S, MAURIN G. Unconventional mechanical and thermal behaviours of MOF CALF-20 [J]. Nature Communications, 2024, 15(1): 3251. doi: 10.1038/s41467-024-47695-6
|
| [147] |
DENG F L, WU H Y, HE R, et al. Large-scale atomistic simulation of dislocation core structure in face-centered cubic metal with deep potential method [J]. Computational Materials Science, 2023, 218: 111941. doi: 10.1016/j.commatsci.2022.111941
|
| [148] |
LI J, LUO K, AN Q. Atomic structure, stability, and dissociation of dislocations in cadmium telluride [J]. International Journal of Plasticity, 2023, 163: 103552. doi: 10.1016/j.ijplas.2023.103552
|
| [149] |
ZHANG J Y, HUYNH G, DAI F Z, et al. A deep-neural network potential to study transformation-induced plasticity in zirconia [J]. Journal of the European Ceramic Society, 2024, 44(6): 4243–4254. doi: 10.1016/j.jeurceramsoc.2024.01.007
|
| [150] |
ZHU C S, DONG W J, GAO Z H, et al. Deep potential fitting and mechanical properties study of MgAlSi alloy [J]. Computational Materials Science, 2024, 239: 112966. doi: 10.1016/j.commatsci.2024.112966
|
| [151] |
WANG R, MA X X, ZHANG L F, et al. Classical and machine learning interatomic potentials for BCC vanadium [J]. Physical Review Materials, 2022, 6(11): 113603. doi: 10.1103/PhysRevMaterials.6.113603
|
| [152] |
MATUSALEM F, REGOJ S, DE KONING M. Plastic deformation of superionic water ices [J]. Proceedings of the National Academy of Sciences of the United States of America, 2022, 119(45): e2203397119. doi: 10.1073/pnas.2203397119
|
| [153] |
WEN T Q, LIU A W, WANG R, et al. Modelling of dislocations, twins and crack-tips in HCP and BCC Ti [J]. International Journal of Plasticity, 2023, 166: 103644. doi: 10.1016/j.ijplas.2023.103644
|
| [154] |
LI Z, SCANDOLO S. Elasticity and viscosity of hcp iron at Earth’s inner core conditions from machine learning-based large-scale atomistic simulations [J]. Geophysical Research Letters, 2022, 49(24): e2022GL101161. doi: 10.1029/2022GL101161
|
| [155] |
LI Z D, HAN L W, OUYANG T, et al. Thermal and mechanical properties of deep-ultraviolet light sources candidate materials BeGeN2 by machine-learning molecular dynamics simulations [J]. Physical Review Materials, 2025, 9(3): 033804. doi: 10.1103/PhysRevMaterials.9.033804
|
| [156] |
ZHAO Z Q, YI M, GUO W L, et al. General-purpose neural network potential for Ti-Al-Nb alloys towards large-scale molecular dynamics with ab initio accuracy [J]. Physical Review B, 2024, 110(18): 184115. doi: 10.1103/PhysRevB.110.184115
|
| [157] |
YING P H, DONG H K, LIANG T, et al. Atomistic insights into the mechanical anisotropy and fragility of monolayer fullerene networks using quantum mechanical calculations and machine-learning molecular dynamics simulations [J]. Extreme Mechanics Letters, 2023, 58: 101929. doi: 10.1016/j.eml.2022.101929
|
| [158] |
ZHANG G Q, ZHAO S W, QIN H S, et al. A unified strength criterion of diamane grain boundaries [J]. Extreme Mechanics Letters, 2024, 68: 102146. doi: 10.1016/j.eml.2024.102146
|
| [159] |
ZENG Q Y, CHEN B, YU X X, et al. Towards large-scale and spatiotemporally resolved diagnosis of electronic density of states by deep learning [J]. Physical Review B, 2022, 105(17): 174109. doi: 10.1103/PhysRevB.105.174109
|
| [160] |
ZENG Q Y, CHEN B, ZHANG S, et al. Full-scale ab initio simulations of laser-driven atomistic dynamics [J]. NPJ Computational Materials, 2023, 9(1): 213. doi: 10.1038/s41524-023-01168-4
|
| [161] |
FU X, WOOD B M, BARROSO-LUQUE L, et al. Learning smooth and expressive interatomic potentials for physical property prediction [C]//Proceedings of the 42nd International Conference on Machine Learning. PMLR, 2025: 17875–17893.
|
| [162] |
YANG H, HU C X, ZHOU Y C, et al. MatterSim: a deep learning atomistic model across elements, temperatures and pressures [EB/OL]. arXiv: 2405.04967. (2024-03-10)[2025-08-26]. https://arxiv.org/abs/2405.04967.
|
| [163] |
ZHANG D, PENG A Y, CAI C, et al. A graph neural network for the era of large atomistic models [EB/OL]. arXiv: 2506.01686. (2025-06-02)[2025-08-26]. https://arxiv.org/abs/2506.01686.
|
| [164] |
BATATIA I, BENNER P, CHIANG Y, et al. A foundation model for atomistic materials chemistry [EB/OL]. arXiv: 2401.00096. (2025-09-04)[2025-08-26]. https://arxiv.org/abs/2401.00096.
|
| [165] |
LIANG T, XU K, LINDGREN E, et al. NEP89: universal neuroevolution potential for inorganic and organic materials across 89 elements [EB/OL]. arXiv: 2504.21286. (2025-06-10)[2025-08-26]. https://arxiv.org/abs/2504.21286.
|
| [166] |
ZHANG L F, WANG H, MUNIZ M C, et al. A deep potential model with long-range electrostatic interactions [J]. The Journal of Chemical Physics, 2022, 156(12): 124107. doi: 10.1063/5.0083669
|
| [167] |
CHENG B Q. Latent Ewald summation for machine learning of long-range interactions [J]. NPJ Computational Materials, 2025, 11(1): 80. doi: 10.1038/s41524-025-01577-7
|
| [168] |
XIA J F, ZHANG Y L, JIANG B. The evolution of machine learning potentials for molecules, reactions and materials [J]. Chemical Society Reviews, 2025, 54(10): 4790–4821. doi: 10.1039/D5CS00104H
|
| [169] |
YANG T, CAI Z F, HUANG Z T, et al. Deep learning illuminates spin and lattice interaction in magnetic materials [J]. Physical Review B, 2024, 110(6): 064427. doi: 10.1103/PhysRevB.110.064427
|
| [170] |
NOVIKOV I, GRABOWSKI B, KÖRMANN F, et al. Magnetic moment tensor potentials for collinear spin-polarized materials reproduce different magnetic states of bcc Fe [J]. NPJ Computational Materials, 2022, 8(1): 13. doi: 10.1038/s41524-022-00696-9
|
| [171] |
KOTYKHOV A S, HODAPP M, TANTARDINI C, et al. Actively trained magnetic moment tensor potentials for mechanical, dynamical, and thermal properties of paramagnetic CrN [J]. Physical Review B, 2025, 111(9): 094438. doi: 10.1103/PhysRevB.111.094438
|
| [172] |
ZHANG Y Z, GAO C, LIU Q R, et al. Warm dense matter simulation via electron temperature dependent deep potential molecular dynamics [J]. Physics of Plasmas, 2020, 27(12): 122704. doi: 10.1063/5.0023265
|
| [173] |
SRINIVASAN P, DEMURIYA D, GRABOWSKI B, et al. Electronic moment tensor potentials include both electronic and vibrational degrees of freedom [J]. NPJ Computational Materials, 2024, 10(1): 41. doi: 10.1038/s41524-024-01222-9
|
| [174] |
LIU Q R, LI J Y, CHEN M H. Thermal transport by electrons and ions in warm dense aluminum: a combined density functional theory and deep potential study [J]. Matter and Radiation at Extremes, 2021, 6(2): 026902. doi: 10.1063/5.0030123
|
| [175] |
HINTON G, VINYALS O, DEAN J. Distilling the knowledge in a neural network [EB/OL]. arXiv: 1503.02531. (2025-03-09)[2025-08-26]. https://arxiv.org/abs/1503.02531.
|
| [176] |
GARDNER J L A, DU TOIT D F T, MAHMOUD C B, et al. Distillation of atomistic foundation models across architectures and chemical domains [EB/OL]. arXiv: 2506.10956. (2025-06-12)[2025-08-26]. https://arxiv.org/abs/2506.10956.
|
Figures(12) / Tables(4)
Website Copyright© Editorial Office of Chinese Journal of High Pressure Physics
Tel:0816-2490042 E-mail:gaoya@caep.cn
Supported by: Beijing Renhe Information Technology Co. Ltd support:
info@rhhz.net








 DownLoad:
DownLoad: