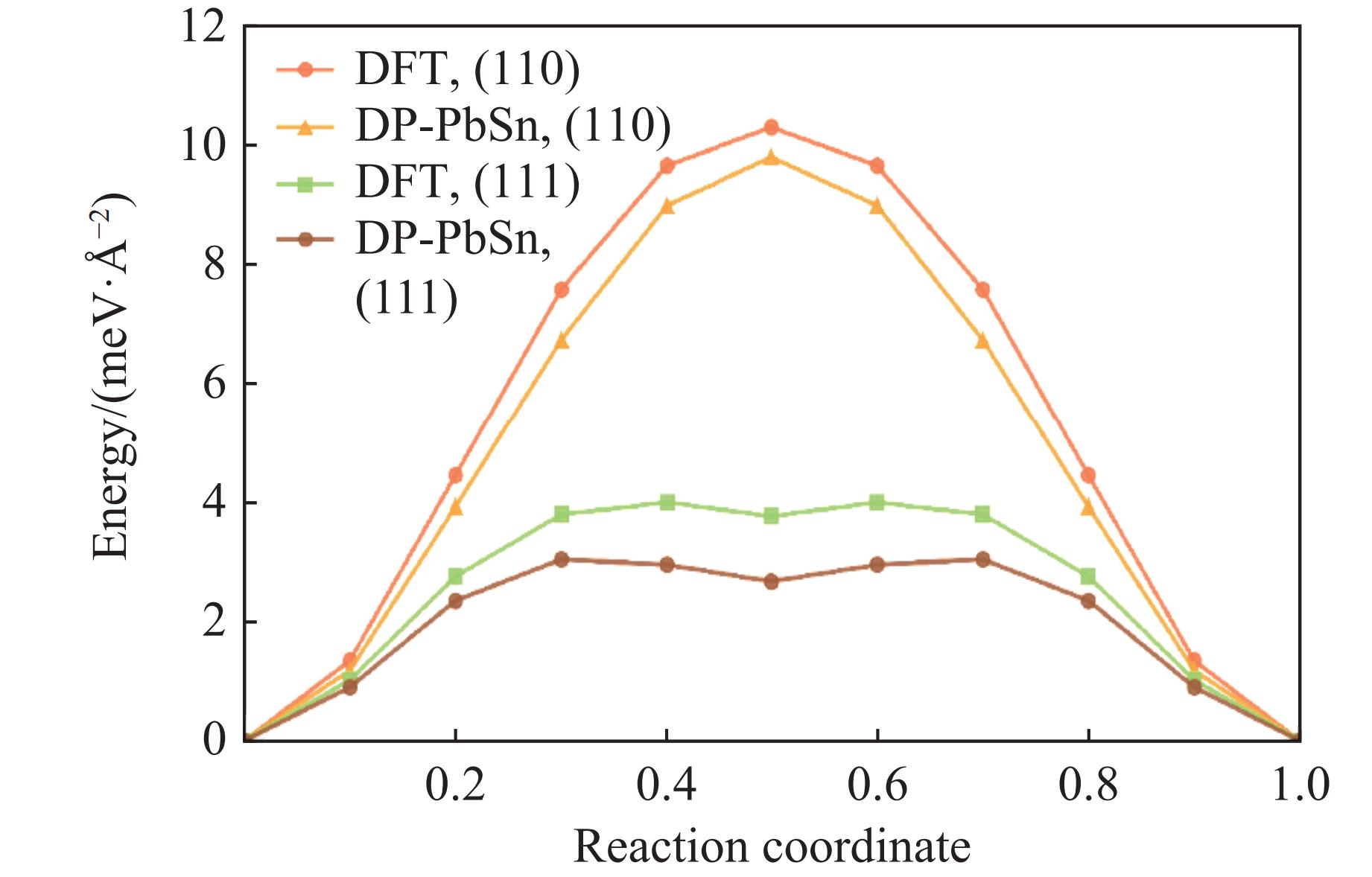
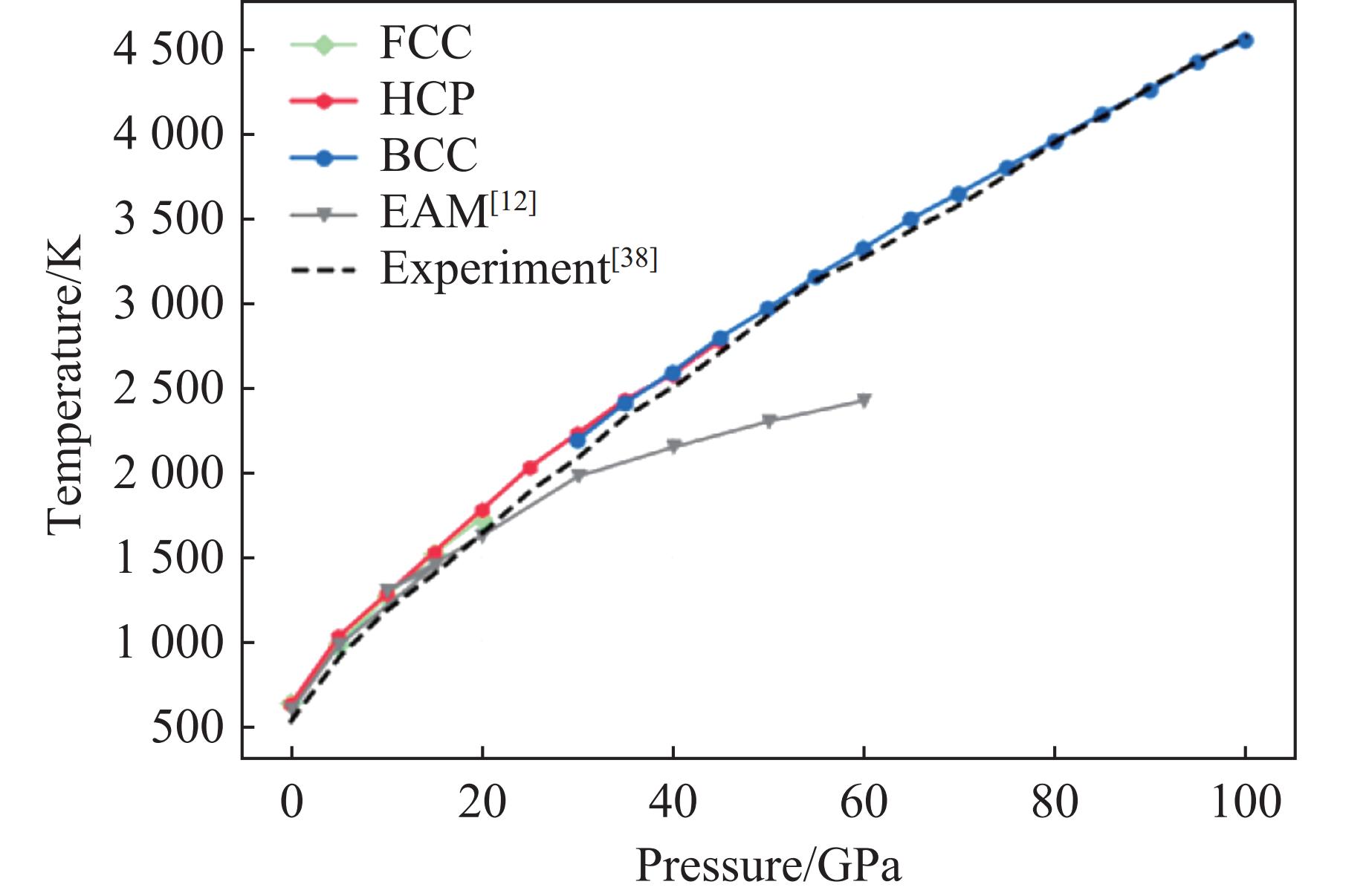
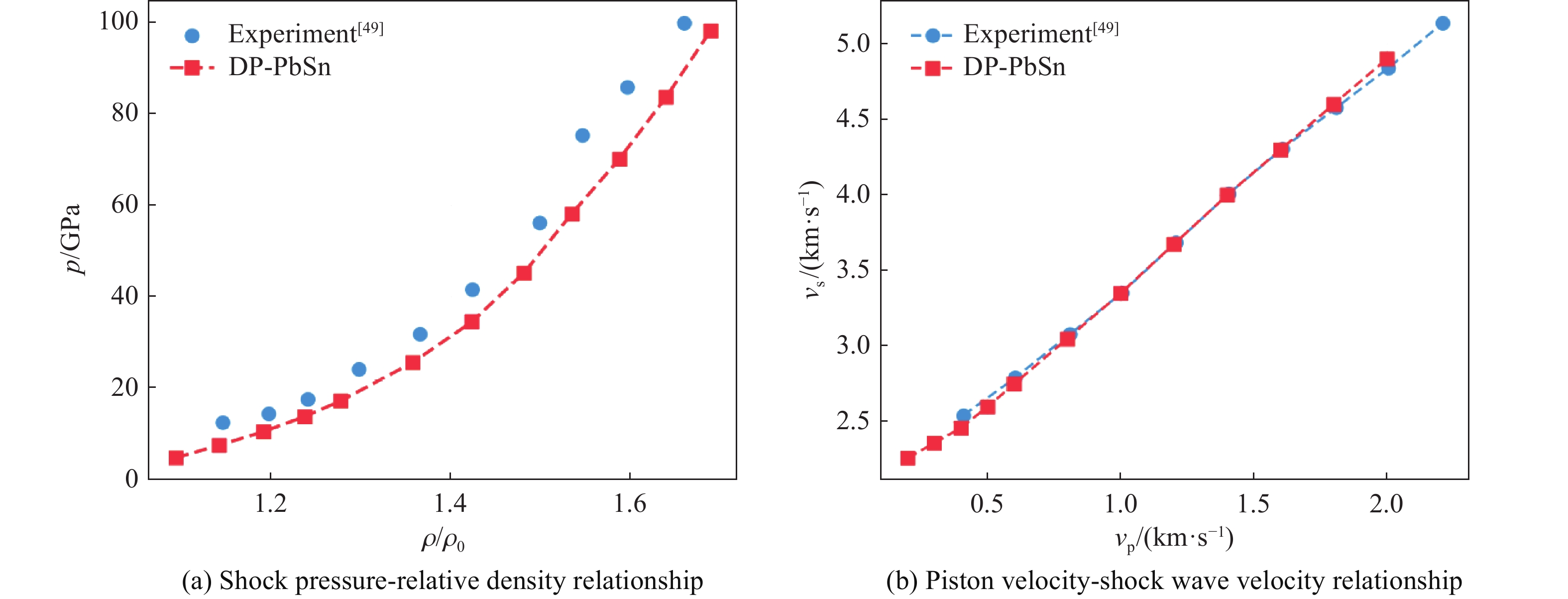
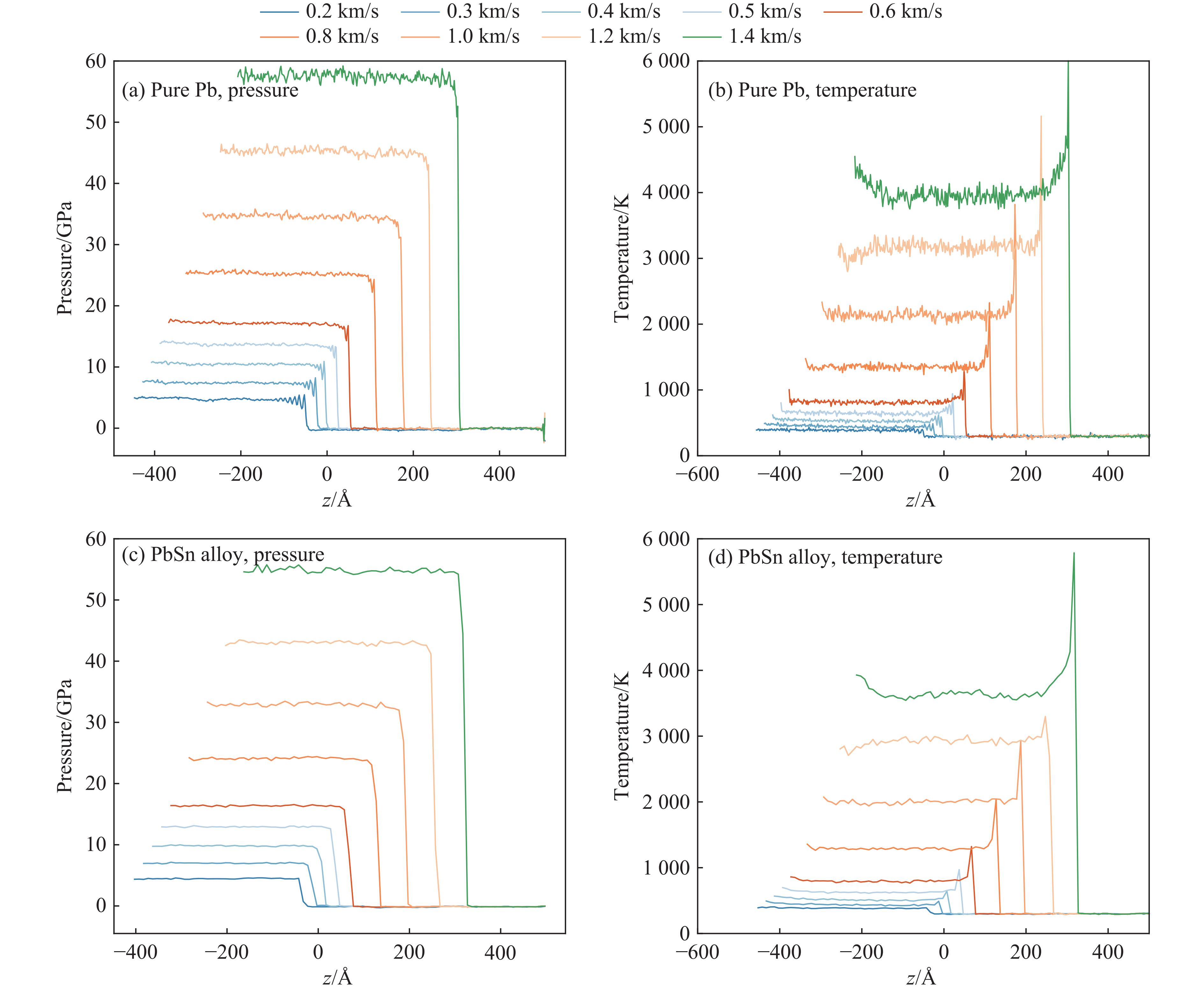
| Citation: | HOU Enze, WANG Xiaoyang, WANG Han. A Machine Learning Potential Model for Simulating Dynamic Mechanical Response of Pb-Sn Alloy[J]. Chinese Journal of High Pressure Physics, 2026, 40(1): 010106. doi: 10.11858/gywlxb.20251151

|

| [1] |
DEWAELE A, MEZOUAR M, GUIGNOT N, et al. Melting of lead under high pressure studied using second-scale time-resolved X-ray diffraction [J]. Physical Review B, 2007, 76(14): 144106. doi: 10.1103/PhysRevB.76.144106
|
| [2] |
KUZNETSOV A, DMITRIEV V, DUBROVINSKY L, et al. FCC-HCP phase boundary in lead [J]. Solid State Communications, 2002, 122(3/4): 125–127. doi: 10.1016/S0038-1098(02)00112-6
|
| [3] |
TAKAHASHI T, MAO H K, BASSETT W A. Lead: X-ray diffraction study of a high-pressure polymorph [J]. Science, 1969, 165(3900): 1352–1353. doi: 10.1126/science.165.3900.1352
|
| [4] |
VANDERBORGH C A, VOHRA Y K, XIA H, et al. BCC lead at 109 GPa: diffraction studies to 208 GPa [J]. Physical Review B, 1990, 41(10): 7338–7340. doi: 10.1103/PhysRevB.41.7338
|
| [5] |
DAI C D, TAN H, GENG H Y. Model for assessing the melting on Hugoniots of metals: Al, Pb, Cu, Mo, Fe, and U [J]. Journal of Applied Physics, 2002, 92(9): 5019–5026. doi: 10.1063/1.1510561
|
| [6] |
KARAKAYA I, THOMPSON W T. The Pb-Sn (lead-tin) system [J]. Journal of Phase Equilibria, 1988, 9(2): 144–152. doi: 10.1007/BF02890552
|
| [7] |
CHEN Y, REN G, TANG T, et al. Experimental study of micro-spalling fragmentation from melted lead [J]. Shock Waves, 2016, 26(2): 221–225. doi: 10.1007/s00193-015-0601-4
|
| [8] |
ANTIPOV M V, ARININ V A, GEORGIEVSKAYA A B, et al. Experimental and computational damage and ejecta studies of Pb explosively shock loaded to pSL≈32 to 40 GPa [J]. Journal of Dynamic Behavior of Materials, 2017, 3(2): 300–315. doi: 10.1007/s40870-017-0113-7
|
| [9] |
XIANG M Z, HU H B, CHEN J. Spalling and melting in nanocrystalline Pb under shock loading: molecular dynamics studies [J]. Journal of Applied Physics, 2013, 113(14): 144312. doi: 10.1063/1.4799388
|
| [10] |
WANG K, ZHANG F G, HE A M, et al. An atomic view on spall responses of release melted lead induced by decaying shock loading [J]. Journal of Applied Physics, 2019, 125(15): 155107. doi: 10.1063/1.5081920
|
| [11] |
MAYER A E, MAYER P N. Strain rate dependence of spall strength for solid and molten lead and tin [J]. International Journal of Fracture, 2020, 222(1/2): 171–195. doi: 10.1007/s10704-020-00440-8
|
| [12] |
LI G M, WANG Y B, WANG K, et al. Shock induced plasticity and phase transition in single crystal lead by molecular dynamics simulations [J]. Journal of Applied Physics, 2019, 126(7): 075902. doi: 10.1063/1.5097621
|
| [13] |
DAW M S, BASKES M I. Embedded-atom method: derivation and application to impurities, surfaces, and other defects in metals [J]. Physical Review B, 1984, 29(12): 6443–6453. doi: 10.1103/PhysRevB.29.6443
|
| [14] |
BASKES M I. Modified embedded-atom potentials for cubic materials and impurities [J]. Physical Review B, 1992, 46(5): 2727–2742. doi: 10.1103/PhysRevB.46.2727
|
| [15] |
HORSFIELD A P, BRATKOVSKY A M, FEARN M, et al. Bond-order potentials: theory and implementation [J]. Physical Review B, 1996, 53(19): 12694–12712. doi: 10.1103/PhysRevB.53.12694
|
| [16] |
WANG X Y, WANG Z Y, GAO P Y, et al. Data-driven prediction of complex crystal structures of dense lithium [J]. Nature Communications, 2023, 14(1): 2924. doi: 10.1038/s41467-023-38650-y
|
| [17] |
WANG K, ZHU W J, XIANG M Z, et al. Improved embedded-atom model potentials of Pb at high pressure: application to investigations of plasticity and phase transition under extreme conditions [J]. Modelling and Simulation in Materials Science and Engineering, 2019, 27(1): 015001. doi: 10.1088/1361-651X/aaea55
|
| [18] |
LIU A Y, GARCA A, COHEN M L, et al. Theory of high-pressure phases of Pb [J]. Physical Review B, 1991, 43(2): 1795–1798. doi: 10.1103/PhysRevB.43.1795
|
| [19] |
CUI S X, CAI L C, FENG W X, et al. First-principles study of phase transition of tin and lead under high pressure [J]. Physica Status Solidi (B), 2008, 245(1): 53–57. doi: 10.1002/pssb.200743240
|
| [20] |
CRICCHIO F, BELONOSHKO A B, BURAKOVSKY L, et al. High-pressure melting of lead [J]. Physical Review B, 2006, 73(14): 140103. doi: 10.1103/PhysRevB.73.140103
|
| [21] |
VOHRA Y K, RUOFF A L. Static compression of metals Mo, Pb, and Pt to 272 GPa: comparison with shock data [J]. Physical Review B, 1990, 42(13): 8651–8654. doi: 10.1103/PhysRevB.42.8651
|
| [22] |
YU D K, SCHEFFLER M. First-principles study of low-index surfaces of lead [J]. Physical Review B, 2004, 70(15): 155417. doi: 10.1103/PhysRevB.70.155417
|
| [23] |
BOMBIS C, EMUNDTS A, NOWICKI M, et al. Absolute surface free energies of Pb [J]. Surface Science, 2002, 511(1/2/3): 83–96. doi: 10.1016/S0039-6028(02)01554-6
|
| [24] |
BARTÓK A P, PAYNE M C, KONDOR R, et al. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons [J]. Physical Review Letters, 2010, 104(13): 136403. doi: 10.1103/PhysRevLett.104.136403
|
| [25] |
SHAPEEV A V. Moment tensor potentials: a class of systematically improvable interatomic potentials [J]. Multiscale Modeling & Simulation, 2016, 14(3): 1153–1173. doi: 10.1137/15M1054183
|
| [26] |
THOMPSON A P, SWILER L P, TROTT C R, et al. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials [J]. Journal of Computational Physics, 2015, 285: 316–330. doi: 10.1016/j.jcp.2014.12.018
|
| [27] |
ZHANG L F, HAN J Q, WANG H, et al. End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems [C]//Proceedings of the 32nd International Conference on Neural Information Processing Systems. Montréal: Curran Associates Inc., 2018: 4441–4451.
|
| [28] |
高天雨, 曾启昱, 陈博, 等. 超快激光诱导Cu薄膜熔化的神经网络分子动力学研究 [J]. 金属学报, 2024, 60(10): 1439–1450. doi: 10.11900/0412.1961.2024.00143
GAO T Y, ZENG Q Y, CHEN B, et al. Neural network molecular dynamics study of ultrafast laser-induced melting of copper nanofilms [J]. Acta Metallurgica Sinica, 2024, 60(10): 1439–1450. doi: 10.11900/0412.1961.2024.00143
|
| [29] |
曾启昱, 陈博, 康冬冬, 等. 大规模、量子精度的分子动力学模拟: 以极端条件液态铁为例 [J]. 物理学报, 2023, 72(18): 187102. doi: 10.7498/aps.72.20231258
ZENG Q Y, CHEN B, KANG D D, et al. Large scale and quantum accurate molecular dynamics simulation: liquid iron under extreme condition [J]. Acta Physica Sinica, 2023, 72(18): 187102. doi: 10.7498/aps.72.20231258
|
| [30] |
D’SOUZA J X, HU S X, MIHAYLOV D I, et al. Designing a quantum-accurate machine-learning potential to enable large-scale simulations of deuterium under shock [J]. Physics of Plasmas, 2025, 32(4): 042701. doi: 10.1063/5.0254638
|
| [31] |
PAN S N, SHI J Y, LIANG Z X, et al. Shock compression pathways to pyrite silica from machine learning simulations [J]. Physical Review B, 2024, 110(22): 224101. doi: 10.1103/PhysRevB.110.224101
|
| [32] |
ZENG Q Y, CHEN B, ZHANG S, et al. Full-scale ab initio simulations of laser-driven atomistic dynamics [J]. NPJ Computational Materials, 2023, 9(1): 213. doi: 10.1038/s41524-023-01168-4
|
| [33] |
ZHANG Y Z, WANG H D, CHEN W J, et al. DP-GEN: a concurrent learning platform for the generation of reliable deep learning based potential energy models [J]. Computer Physics Communications, 2020, 253: 107206. doi: 10.1016/j.cpc.2020.107206
|
| [34] |
WANG H, ZHANG L F, HAN J Q, et al. DeePMD-kit: a deep learning package for many-body potential energy representation and molecular dynamics [J]. Computer Physics Communications, 2018, 228: 178–184. doi: 10.1016/j.cpc.2018.03.016
|
| [35] |
KINGMA D P, BA J. Adam: a method for stochastic optimization [C]//Proceedings of the 3rd International Conference on Learning Representations (ICLR). San Diego: ICLR, 2015.
|
| [36] |
NOSÉ S. A unified formulation of the constant temperature molecular dynamics methods [J]. The Journal of Chemical Physics, 1984, 81(1): 511–519. doi: 10.1063/1.447334
|
| [37] |
HOOVER W G. Canonical dynamics: equilibrium phase-space distributions [J]. Physical Review A, 1985, 31(3): 1695–1697. doi: 10.1103/PhysRevA.31.1695
|
| [38] |
KRYGIER A, POWELL P D, MCNANEY J M, et al. Extreme hardening of Pb at high pressure and strain rate [J]. Physical Review Letters, 2019, 123(20): 205701. doi: 10.1103/PhysRevLett.123.205701
|
| [39] |
ERNZERHOF M, SCUSERIA G E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional [J]. The Journal of Chemical Physics, 1999, 110(11): 5029–5036. doi: 10.1063/1.478401
|
| [40] |
SCHLIPF M, GYGI F. Optimization algorithm for the generation of ONCV pseudopotentials [J]. Computer Physics Communications, 2015, 196: 36–44. doi: 10.1016/j.cpc.2015.05.011
|
| [41] |
ZHOU W Q, ZHENG D Y, LIU Q R, et al. ABACUS: an electronic structure analysis package for the AI era [EB/OL]. arXiv: 2501.08697, 2025 (2025-10-22)[2025-08-06]. https://arxiv.org/abs/2501.08697.
|
| [42] |
CHEN T, YUAN F B, LIU J C, et al. Modeling the high-pressure solid and liquid phases of tin from deep potentials with ab initio accuracy [J]. Physical Review Materials, 2023, 7(5): 053603. doi: 10.1103/PhysRevMaterials.7.053603
|
| [43] |
LI W, LU S, HU Q M, et al. Generalized stacking fault energies of alloys [J]. Journal of Physics: Condensed Matter, 2014, 26(26): 265005. doi: 10.1088/0953-8984/26/26/265005
|
| [44] |
余永宁, 杨平, 强文江, 等. 材料科学基础 [M]. 北京: 高等教育出版社, 2006.
YU Y N, YANG P, QIANG W J, et al. Fundamentals of materials science [M]. Beijing: Higher Education Press, 2006.
|
| [45] |
MO P H, LI C, ZHAO D, et al. Accurate and efficient molecular dynamics based on machine learning and non von Neumann architecture [J]. NPJ Computational Materials, 2022, 8(1): 107. doi: 10.1038/s41524-022-00773-z
|
| [46] |
Sandia National Laboratories. LAMMPS [CP/OL]. [2025-10-02]. https://lammps. sandia. gov.
|
| [47] |
LI W H, HAHN E N, YAO X H, et al. Shock induced damage and fracture in SiC at elevated temperature and high strain rate [J]. Acta Materialia, 2019, 167: 51–70. doi: 10.1016/j.actamat.2018.12.035
|
| [48] |
STUKOWSKI A. Visualization and analysis of atomistic simulation data with OVITO—the open visualization tool [J]. Modelling and Simulation in Materials Science and Engineering, 2010, 18(1): 015012. doi: 10.1088/0965-0393/18/1/015012
|
| [49] |
MARSH S P. LASL shock Hugoniot data [M]. Berkeley: University of California Press, 1980.
|
| [50] |
ALIPPI P, MARCUS P M, SCHEFFLER M. Strained tetragonal states and Bain paths in metals [J]. Physical Review Letters, 1997, 78(20): 3892–3895. doi: 10.1103/PhysRevLett.78.3892
|
Figures(6) / Tables(3)
Website Copyright© Editorial Office of Chinese Journal of High Pressure Physics
Tel:0816-2490042 E-mail:gaoya@caep.cn
Supported by: Beijing Renhe Information Technology Co. Ltd support:
info@rhhz.net








 DownLoad:
DownLoad: