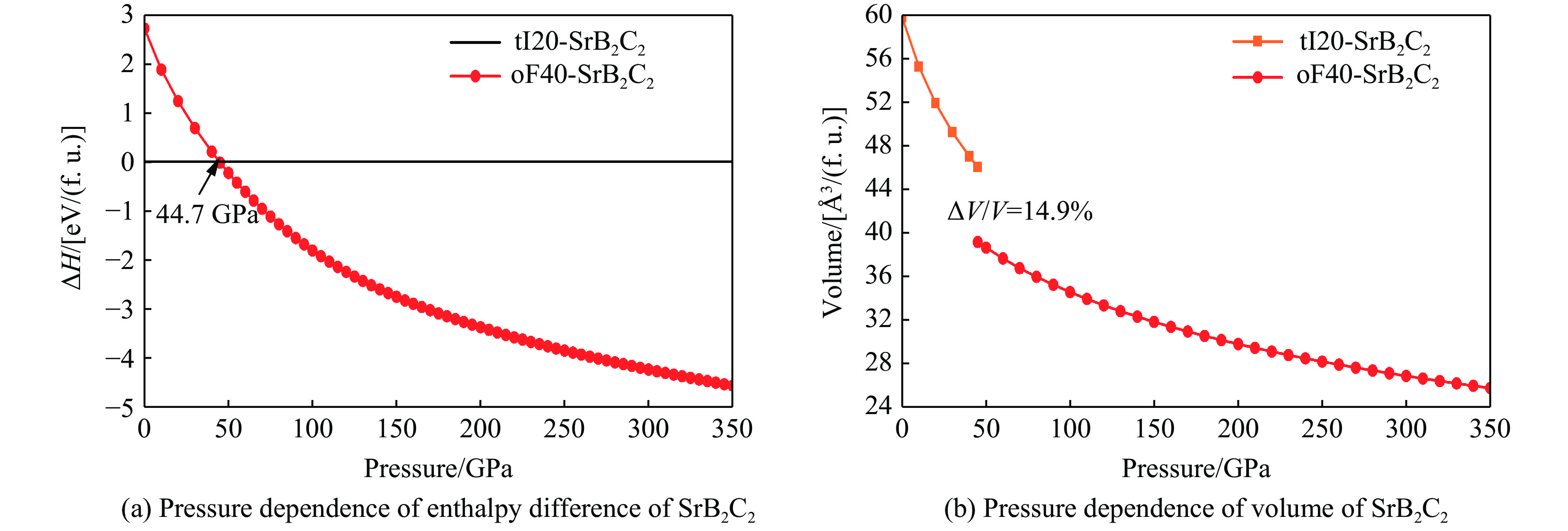
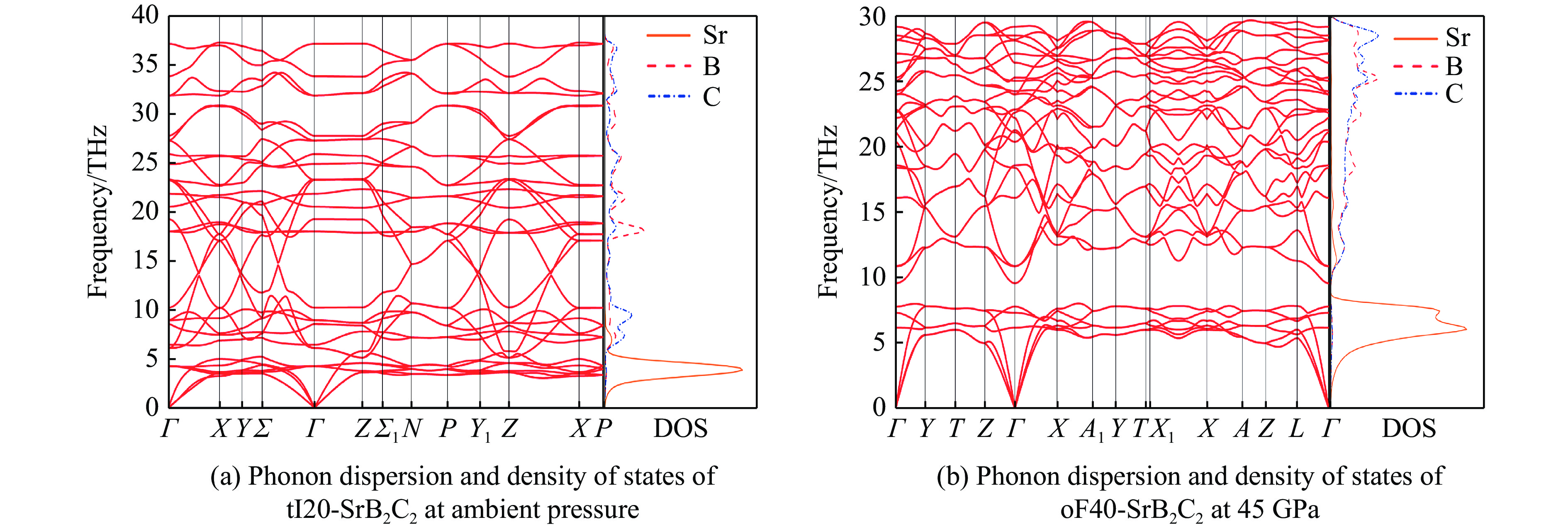
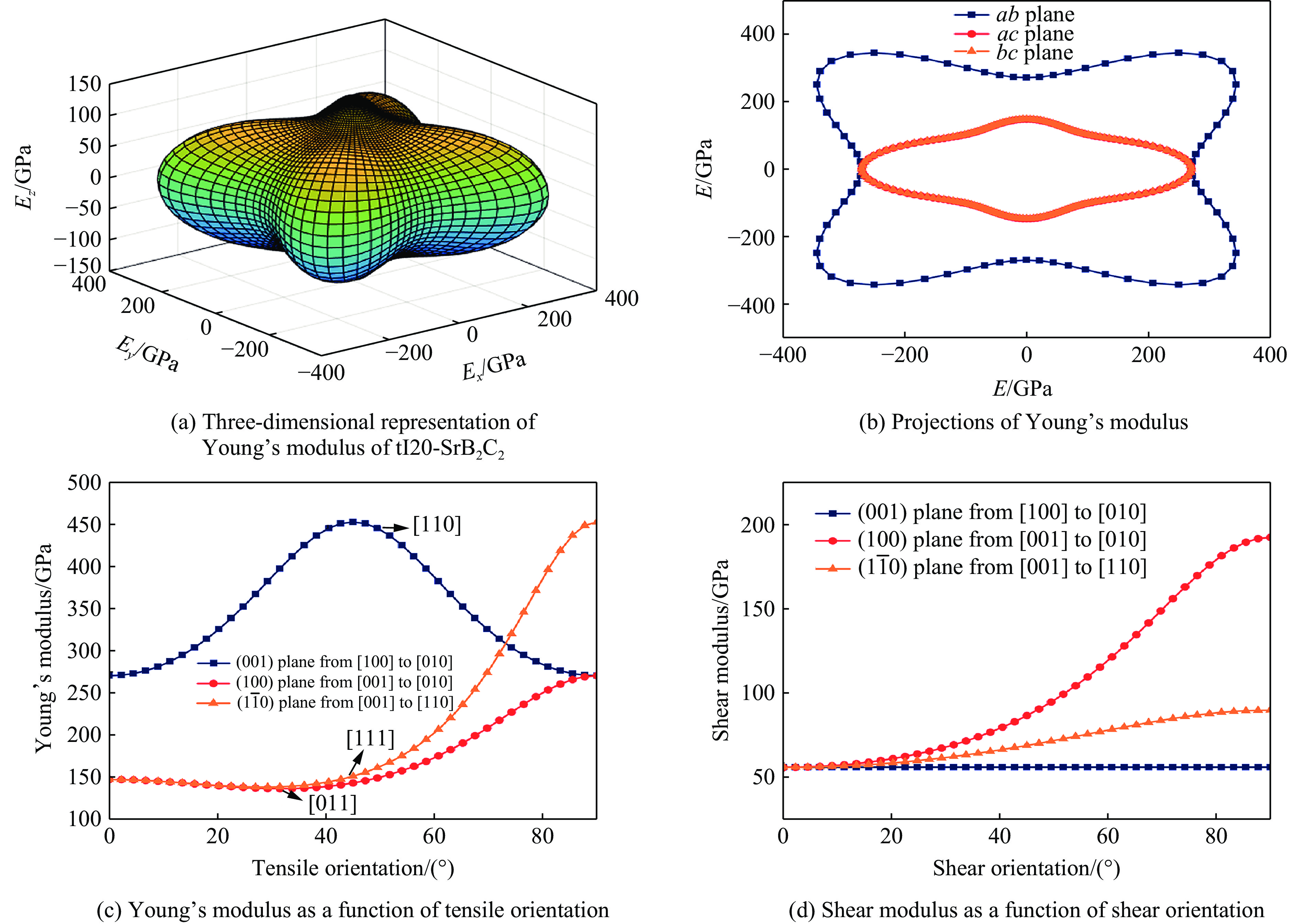
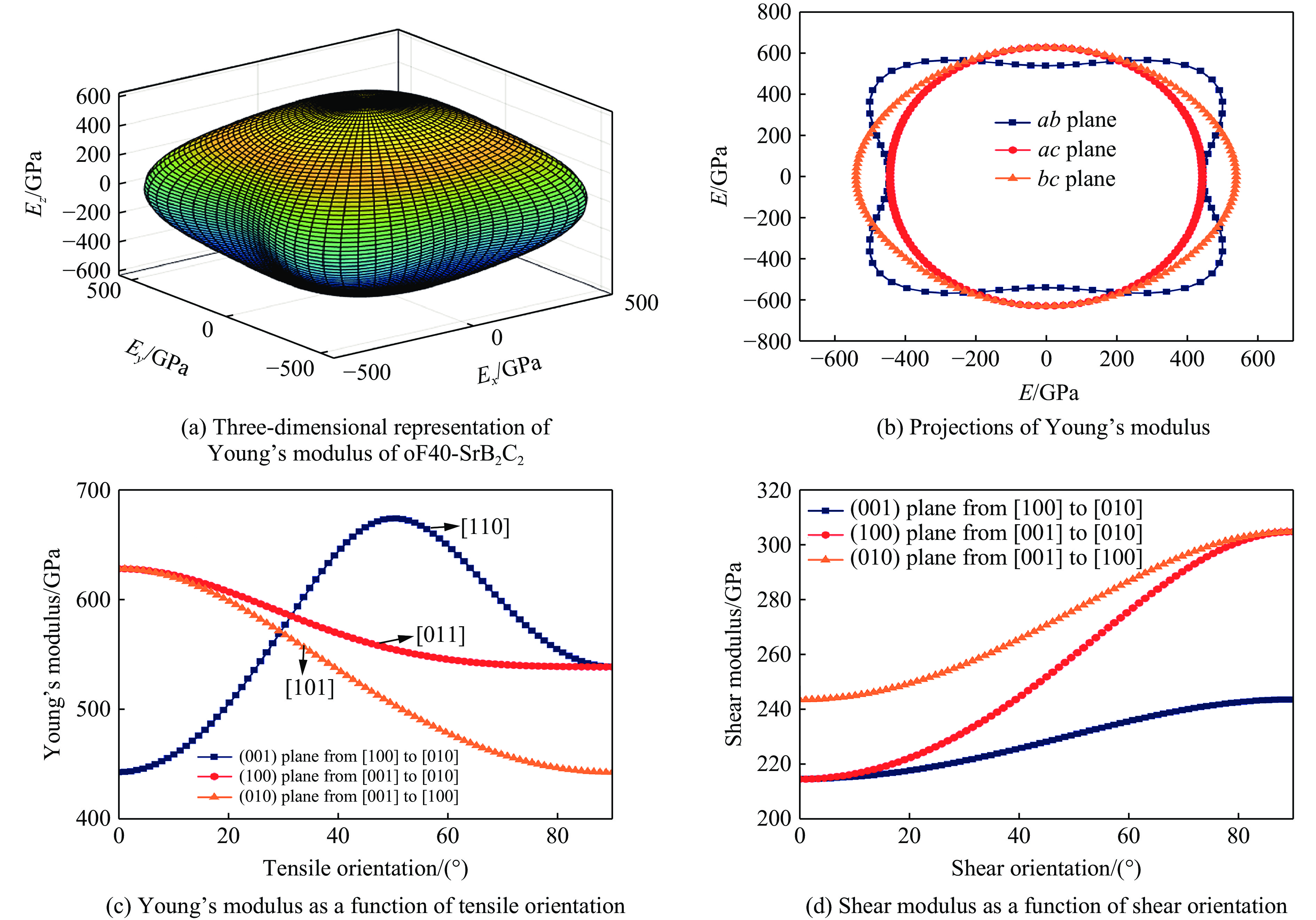
| Citation: | GUO Hua, WANG Fan, ZHENG Baobing. High-Pressure Study on Structural Phase Transformation and Physical Properties of SrB2C2[J]. Chinese Journal of High Pressure Physics, 2026, 40(1): 010108. doi: 10.11858/gywlxb.20251148

|

| [1] |
NAGAMATSU J, NAKAGAWA N, MURANAKA T, et al. Superconductivity at 39 K in magnesium diboride [J]. Nature, 2001, 410(6824): 63–64. doi: 10.1038/35065039
|
| [2] |
SHAH S, KOLMOGOROV A N. Stability and superconductivity of Ca-B phases at ambient and high pressure [J]. Physical Review B, 2013, 88(1): 014107. doi: 10.1103/PhysRevB.88.014107
|
| [3] |
EMERY N, HÉROLD C, D’ASTUTO M, et al. Superconductivity of bulk CaC6 [J]. Physical Review Letters, 2005, 95(8): 087003. doi: 10.1103/PhysRevLett.95.087003
|
| [4] |
GAO M, YAN X W, LU Z Y, et al. Strong-coupling superconductivity in LiB2C2 trilayer films [J]. Physical Review B, 2020, 101(9): 094501. doi: 10.1103/PhysRevB.101.094501
|
| [5] |
ZHENG B B. Pressure-induced phase transition and electronic properties of MgB2C2 [J]. Journal of Applied Physics, 2017, 121(19): 195102. doi: 10.1063/1.4983821
|
| [6] |
YAN H Y, ZHANG M G, WEI Q, et al. Ab initio studies of ternary semiconductor BeB2C2 [J]. Computational Materials Science, 2013, 68: 174–180. doi: 10.1016/j.commatsci.2012.10.013
|
| [7] |
ZHENG B B, ZHANG M G, CHANG S M. Structural, mechanical and electronic properties of CaB2C2 at high pressure [J]. Europhysics Letters, 2017, 118(6): 66001. doi: 10.1209/0295-5075/118/66001
|
| [8] |
AKIMITSU J, TAKENAWA K, SUZUKI K, et al. High-temperature ferromagnetism in CaB2C2 [J]. Science, 2001, 293(5532): 1125–1127. doi: 10.1126/science.1061501
|
| [9] |
HAQUE E, HOSSAIN M A, STAMPFL C. First-principles prediction of phonon-mediated superconductivity in XBC (X=Mg, Ca, Sr, Ba) [J]. Physical Chemistry Chemical Physics, 2019, 21(17): 8767–8773. doi: 10.1039/c8cp07634k
|
| [10] |
ZHU L, BORSTAD G M, LIU H Y, et al. Carbon-boron clathrates as a new class of sp3-bonded framework materials [J]. Science Advances, 2020, 6(2): eaay8361. doi: 10.1126/sciadv.aay8361
|
| [11] |
WANG J N, YAN X W, GAO M. High-temperature superconductivity in SrB3C3 and BaB3C3 predicted from first-principles anisotropic Migdal-Eliashberg theory [J]. Physical Review B, 2021, 103(14): 144515. doi: 10.1103/PhysRevB.103.144515
|
| [12] |
ZHU L, LIU H Y, SOMAYAZULU M, et al. Superconductivity in SrB3C3 clathrate [J]. Physical Review Research, 2023, 5(1): 013012. doi: 10.1103/PhysRevResearch.5.013012
|
| [13] |
ZHANG Y M, CHEN J Y, HAO J, et al. Conventional high-temperature superconductivity in σ-band driven metallized two-dimensional metal borocarbides [J]. Physical Review B, 2024, 110(6): 064513. doi: 10.1103/PhysRevB.110.064513
|
| [14] |
BURDETT J K, LEE S, MCLARNAN T J. Coloring problem [J]. Journal of the American Chemical Society, 1985, 107(11): 3083–3089. doi: 10.1021/ja00297a012
|
| [15] |
WHEELER R A, WHANGBO M H, HUGHBANKS T, et al. Symmetric vs. asymmetric linear M-X-M linkages in molecules, polymers, and extended networks [J]. Journal of the American Chemical Society, 1986, 108(9): 2222–2236. doi: 10.1021/ja00269a018
|
| [16] |
MILLER G J. The “coloring problem” in solids: how it affects structure, composition and properties [J]. European Journal of Inorganic Chemistry, 1998, 1998(5): 523–536. doi: 10.1002/(SICI)1099-0682(199805)1998:5<523::AID-EJIC523>3.0.CO;2-L
|
| [17] |
DOMNICH V, REYNAUD S, HABER R A, et al. Boron carbide: structure, properties, and stability under stress [J]. Journal of the American Ceramic Society, 2011, 94(11): 3605–3628. doi: 10.1111/j.1551-2916.2011.04865.x
|
| [18] |
LONIE D C, ZUREK E. XtalOpt: an open-source evolutionary algorithm for crystal structure prediction [J]. Computer Physics Communications, 2011, 182(2): 372–387. doi: 10.1016/j.cpc.2010.07.048
|
| [19] |
WANG Y C, LV J, ZHU L, et al. Crystal structure prediction via particle-swarm optimization [J]. Physical Review B, 2010, 82(9): 094116. doi: 10.1103/PhysRevB.82.094116
|
| [20] |
WANG Y C, LV J, ZHU L, et al. CALYPSO: a method for crystal structure prediction [J]. Computer Physics Communications, 2012, 183(10): 2063–2070. doi: 10.1016/j.cpc.2012.05.008
|
| [21] |
LU C, LI Q, MA Y M, et al. Extraordinary indentation strain stiffening produces superhard tungsten nitrides [J]. Physical Review Letters, 2017, 119(11): 115503. doi: 10.1103/PhysRevLett.119.115503
|
| [22] |
LU S H, WANG Y C, LIU H Y, et al. Self-assembled ultrathin nanotubes on diamond (100) surface [J]. Nature Communications, 2014, 5(1): 3666. doi: 10.1038/ncomms4666
|
| [23] |
HE X L, ZHAO W B, XIE Y, et al. Predicted hot superconductivity in LaSc2H24 under pressure [J]. Proceedings of the National Academy of Sciences of the United States of America, 2024, 121(26): e2401840121. doi: 10.1073/pnas.2401840121
|
| [24] |
MA C H, MA Y, WANG H, et al. Hydrogen-vacancy-induced stable superconducting niobium hydride at high pressure [J]. Journal of the American Chemical Society, 2025, 147(13): 11028–11035. doi: 10.1021/jacs.4c15868
|
| [25] |
LUO X Y, YANG J H, LIU H Y, et al. Predicting two-dimensional boron-carbon compounds by the global optimization method [J]. Journal of the American Chemical Society, 2011, 133(40): 16285–16290. doi: 10.1021/ja2072753
|
| [26] |
WANG Y, LI F, LI Y F, et al. Semi-metallic Be5C2 monolayer global minimum with quasi-planar pentacoordinate carbons and negative Poisson’s ratio [J]. Nature Communications, 2016, 7(1): 11488. doi: 10.1038/ncomms11488
|
| [27] |
TANG C, KOUR G, DU A J. Recent progress on the prediction of two-dimensional materials using CALYPSO [J]. Chinese Physics B, 2019, 28(10): 107306. doi: 10.1088/1674-1056/ab41ea
|
| [28] |
MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations [J]. Physical Review B, 1976, 13(12): 5188–5192. doi: 10.1103/PhysRevB.13.5188
|
| [29] |
COLLE R, SALVETTI O. Approximate calculation of the correlation energy for the closed shells [J]. Theoretica Chimica Acta, 1975, 37(4): 329–334. doi: 10.1007/BF01028401
|
| [30] |
PERDEW J P, CHEVARY J A, VOSKO S H, et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation [J]. Physical Review B, 1992, 46(11): 6671–6687. doi: 10.1103/PhysRevB.46.6671
|
| [31] |
METHFESSEL M, PAXTON A T. High-precision sampling for Brillouin-zone integration in metals [J]. Physical Review B, 1989, 40(6): 3616–3621. doi: 10.1103/PhysRevB.40.3616
|
| [32] |
NYE J F. Physical properties of crystals: their representation by tensors and matrices [J]. Acta Crystallographica Section A, 1985, 41(6): 624–624.
|
| [33] |
WANG V, XU N, LIU J C, et al. VASPKIT: a user-friendly interface facilitating high-throughput computing and analysis using VASP code [J]. Computer Physics Communications, 2021, 267: 108033. doi: 10.1016/j.cpc.2021.108033
|
| [34] |
MOUHAT F, COUDERT F X. Necessary and sufficient elastic stability conditions in various crystal systems [J]. Physical Review B, 2014, 90(22): 224104. doi: 10.1103/PhysRevB.90.224104
|
| [35] |
方俊鑫, 陆栋. 固体物理学[M]. 上海: 上海科学技术出版社, 1980.
|
| [36] |
CHANDRASEKAR S, SANTHANAM S. A calculation of the bulk modulus of polycrystalline materials [J]. Journal of Materials Science, 1989, 24(12): 4265–4267. doi: 10.1007/BF00544497
|
| [37] |
PANDA K B, CHANDRAN K S R. Determination of elastic constants of titanium diboride (TiB2) from first principles using FLAPW implementation of the density functional theory [J]. Computational Materials Science, 2006, 35(2): 134–150. doi: 10.1016/j.commatsci.2005.03.012
|
| [38] |
YAN H Y, ZHANG M G, WEI Q, et al. Elastic anisotropy and thermodynamic properties of tetrahedrally bonded dense C2N2 (NH) under high pressure and high temperature [J]. Physica Status Solidi B, 2013, 250(7): 1293–1299. doi: 10.1002/pssb.201248225
|
| [39] |
CAZZANI A, ROVATI M. Extrema of Young’s modulus for elastic solids with tetragonal symmetry [J]. International Journal of Solids and Structures, 2005, 42(18/19): 5057–5096. doi: 10.1016/j.ijsolstr.2005.02.018
|
| [40] |
HE Y, SCHWARZ R B, MIGLIORI A, et al. Elastic constants of single crystal γ-TiAl [J]. Journal of Materials Research, 1995, 10(5): 1187–1195. doi: 10.1557/JMR.1995.1187
|
Figures(8) / Tables(2)
Website Copyright© Editorial Office of Chinese Journal of High Pressure Physics
Tel:0816-2490042 E-mail:gaoya@caep.cn
Supported by: Beijing Renhe Information Technology Co. Ltd support:
info@rhhz.net








 DownLoad:
DownLoad: