




Load Characteristics of Underwater Explosion Shock Wave near Seabed Charge Projectile
-
摘要: 近海底爆炸冲击波会对海底光缆、海底管道等设施造成严重的破坏,不同底质条件的冲击阻抗会影响冲击波的时空演化规律,因此,研究不同底质条件下近海底爆炸冲击波载荷具有重要的意义。基于耦合欧拉-拉格朗日方法建立近海底爆炸模型,探究海底底质对近海底爆炸冲击波载荷的影响,结果显示:测点角度在20°~50°范围内时,海底底质材料会显著影响冲击波峰值压力,近海底反射的影响随爆距比的增加而增强,当测点角度超出该范围时,该现象逐渐消失;海底底质影响范围不随底质的变化而变化,但是在不同底质条件下,受影响区域的反射系数截然不同,当海底底质较软时,海底底质影响区域内的冲击波反射系数在0.81~1.05之间,当海底底质为刚壁时,海底底质影响区域内的冲击波反射系数在0.98~1.33之间;水深不会导致冲击波峰值压力发生显著变化。Abstract: The near-seabed explosion shock wave can cause serious damage to such facilities as submarine optical cables and pipelines. The spatiotemporal evolution of the underwater explosion shock wave can be affected by the impedance of different types of substrate. Therefore, it is of great significance to study the near-seabed explosion shock wave load under different substrate conditions. Based on the coupled Eulerian-Lagrangian (CEL) method, a numerical model of near seabed explosion is established to investigate the effect of seabed material on the shock wave load of near seabed explosions. The results show that the seabed material significantly affects the peak pressure of the shock wave within a certain range of measuring point angles of 20°–50°. The reflection effect of the near seabed increases with the increase of the explosion distance ratio within a certain range of measurement point angles of 20°–50°. When the measurement point angle exceeds this range, this phenomenon gradually disappears. The effect regions of these two types of seabed sediment are similar while their reflection coefficients are significantly different. When the seafloor sediment is soft, the reflection coefficient near seafloor ranges from 0.81 to 1.05. However, when the seafloor sediment is hard, the reflection coefficient near seafloor ranges from 0.98 to 1.33. The water depth has little effect on the peak pressure of the shock wave.
-
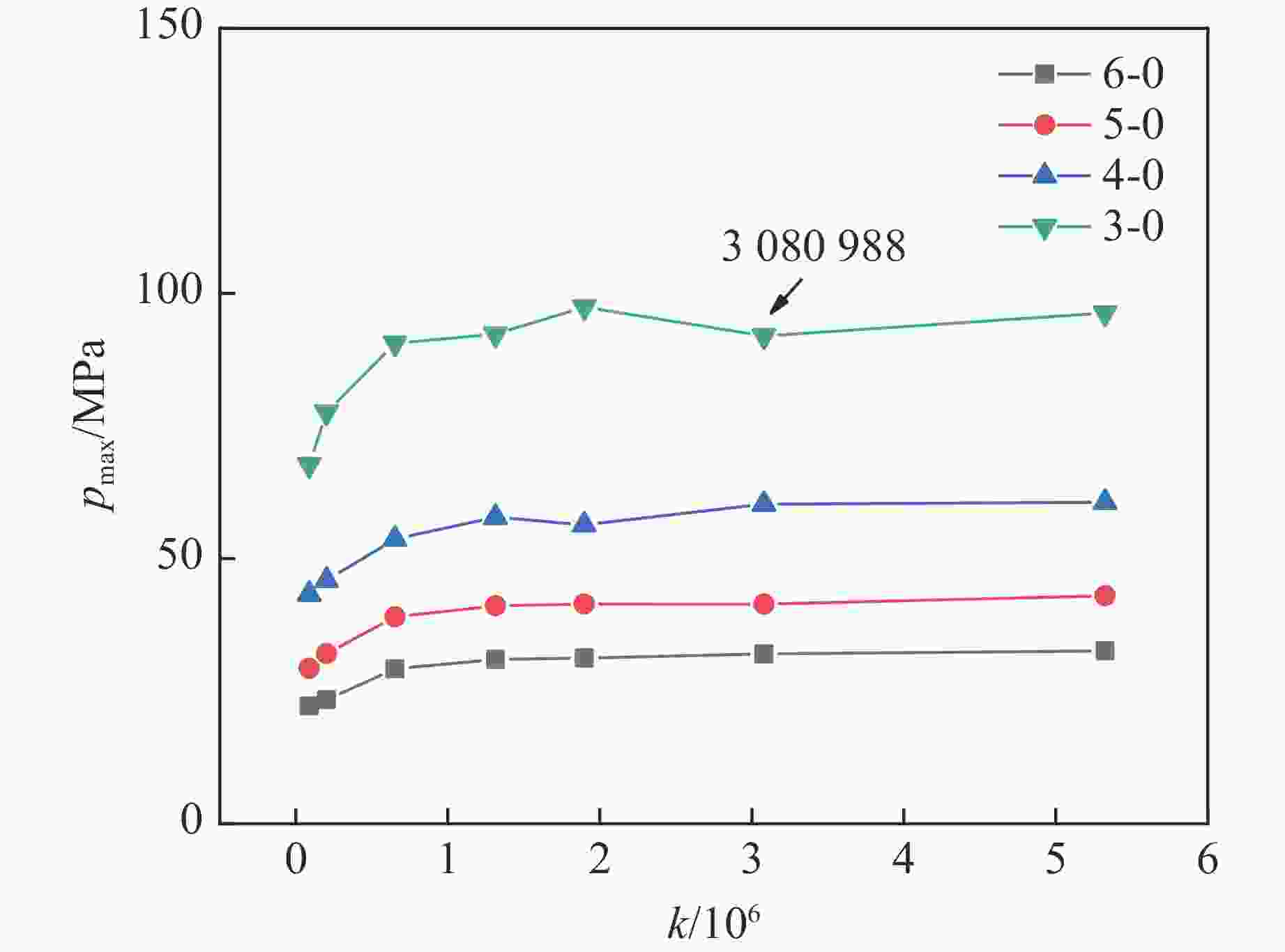
图 2 不同网格数量下不同测点测得的压力峰值
Figure 2. Peak pressures measured at different points and grid numbers
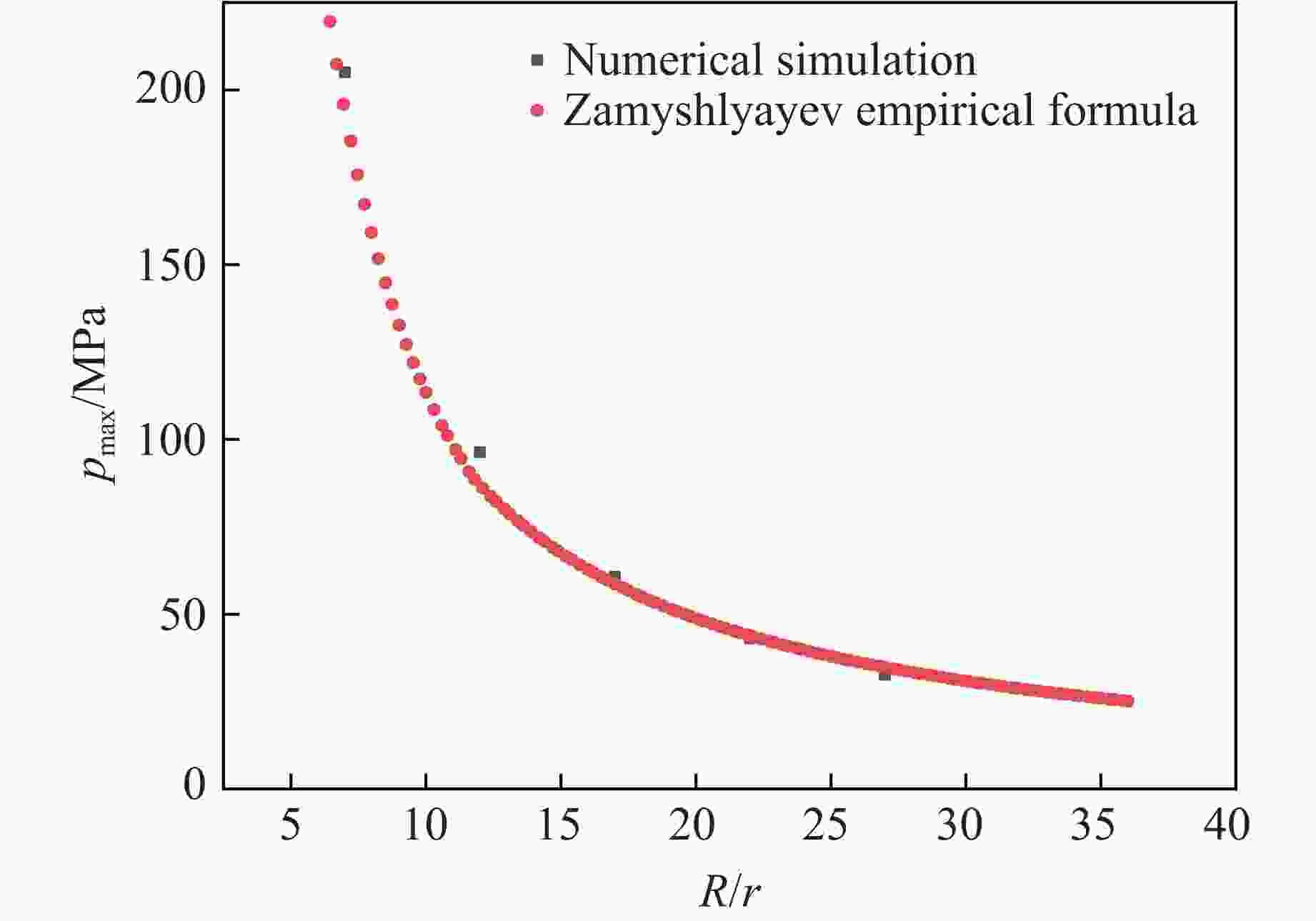
图 3 不同距离处冲击波峰值压力对比
Figure 3. Comparison of peak pressure of shock waves at different distances
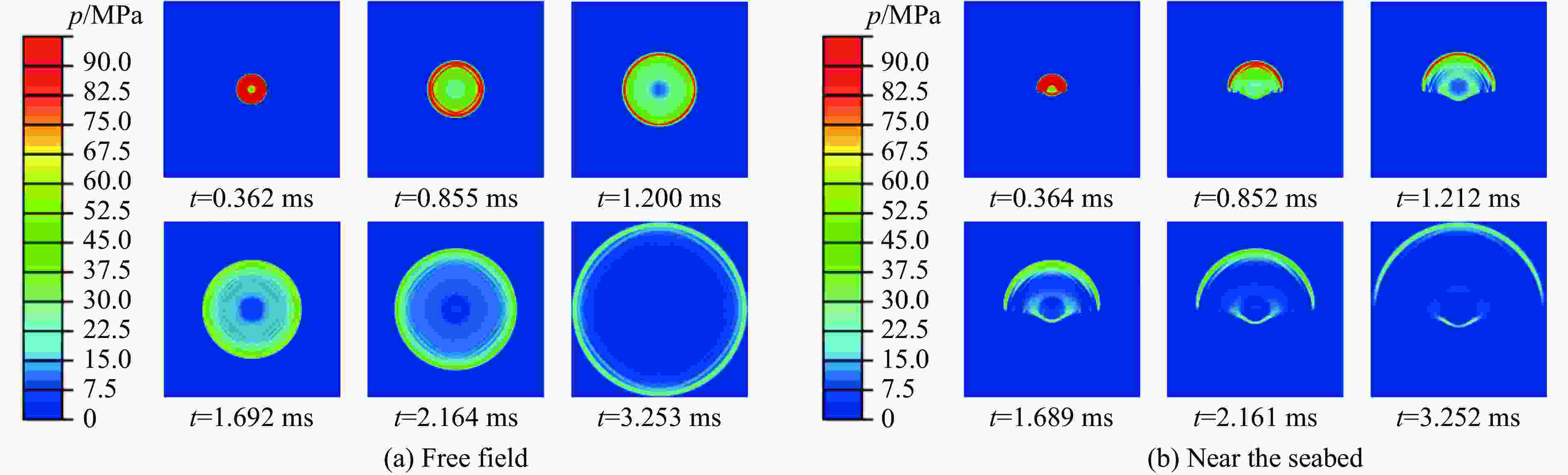
图 4 自由场及近海底水下爆炸冲击波压力云图
Figure 4. Pressure distribution of free-field and near-seabed underwater explosion shock wave
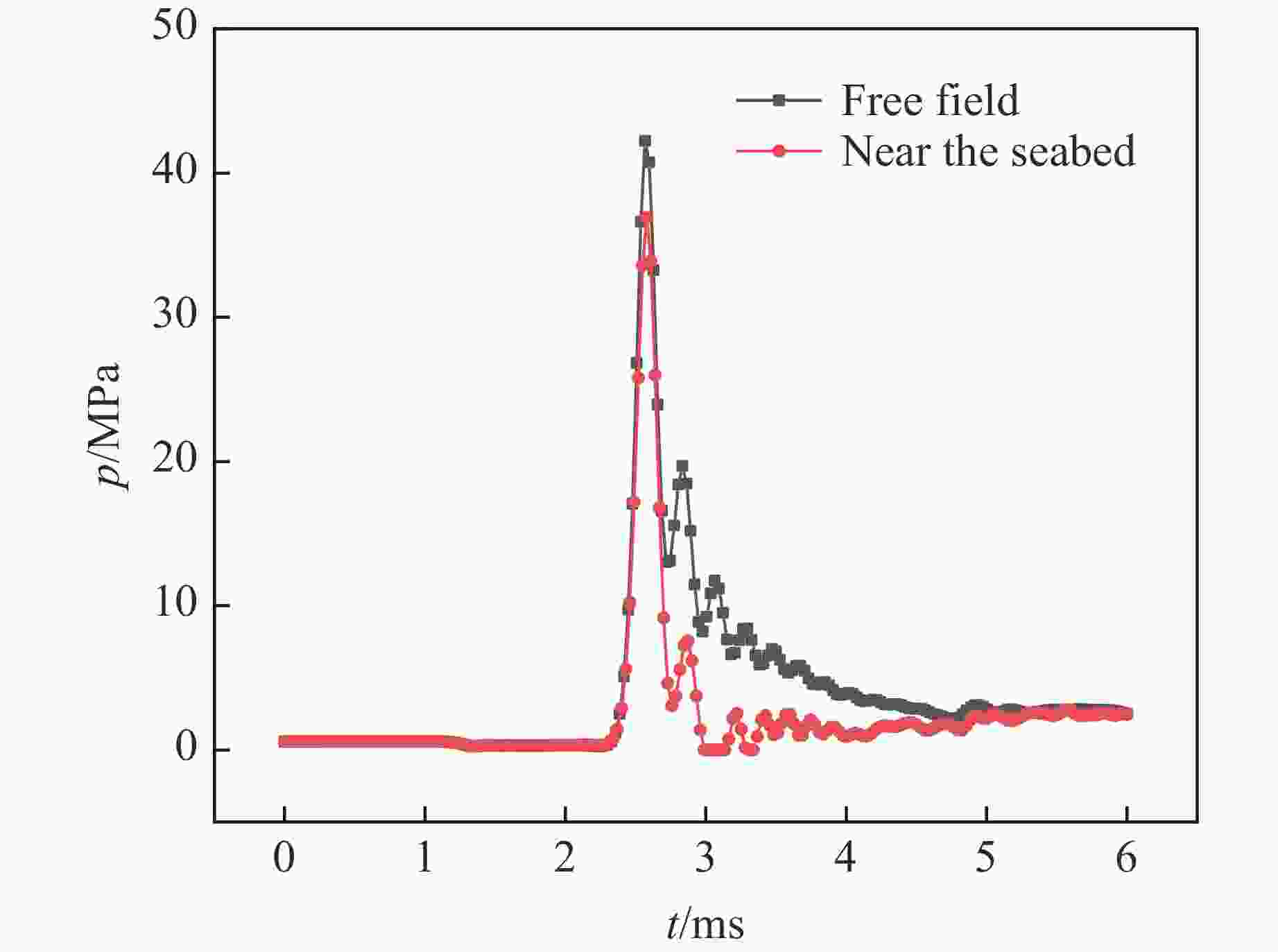
图 5 自由场及近海底工况下测点5-3的水下爆炸冲击波压力时程曲线
Figure 5. Time history curves of shock wave pressure at measuring point 5-3 in free field and near seabed underwater explosion

图 7 不同底质条件下测点反射系数的对比
Figure 7. Comparison of reflection coefficient of test points under different substrate conditions
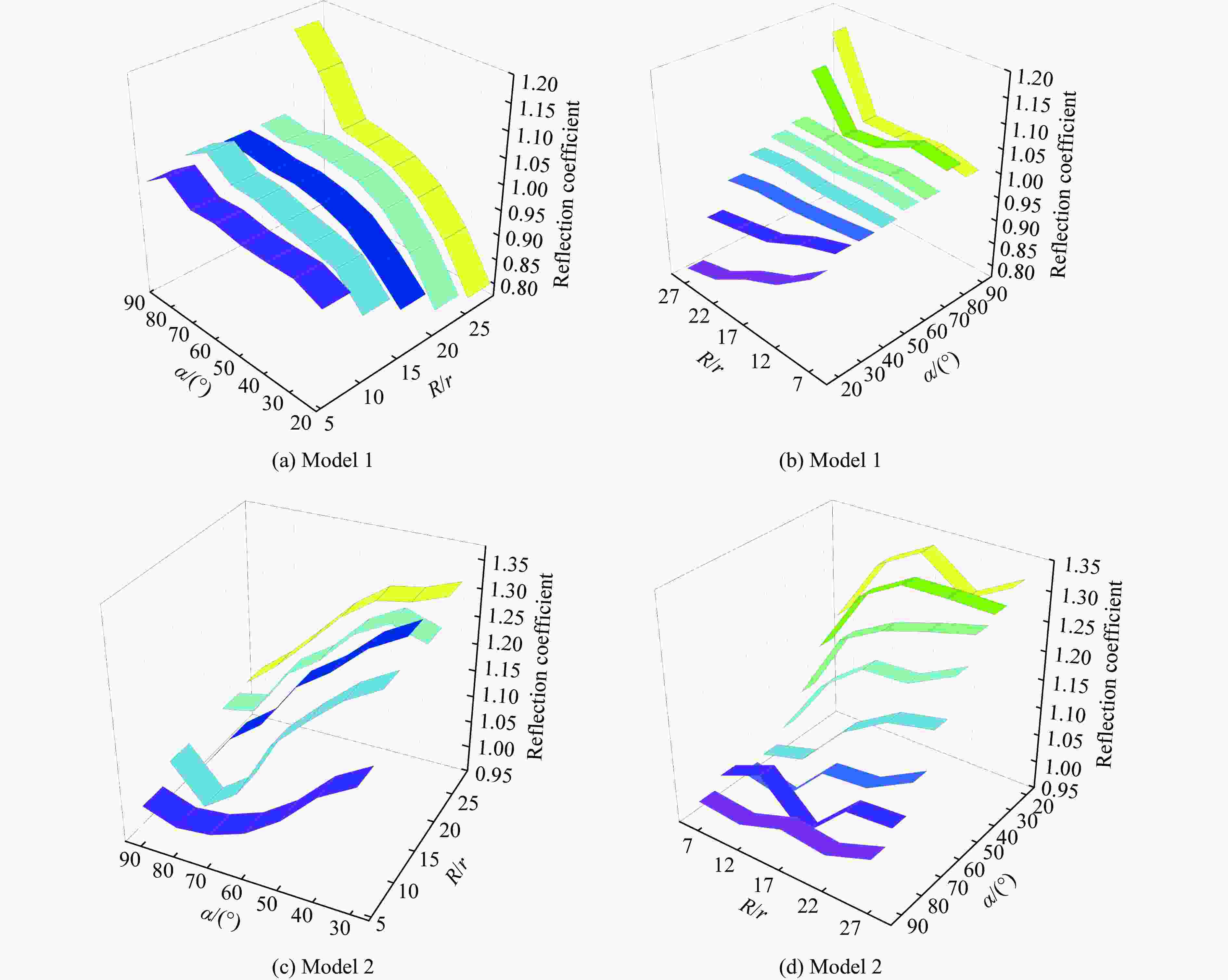
图 8 不同底质条件下反射系数分布对比
Figure 8. Comparison of reflection coefficient distribution under different substrate conditions
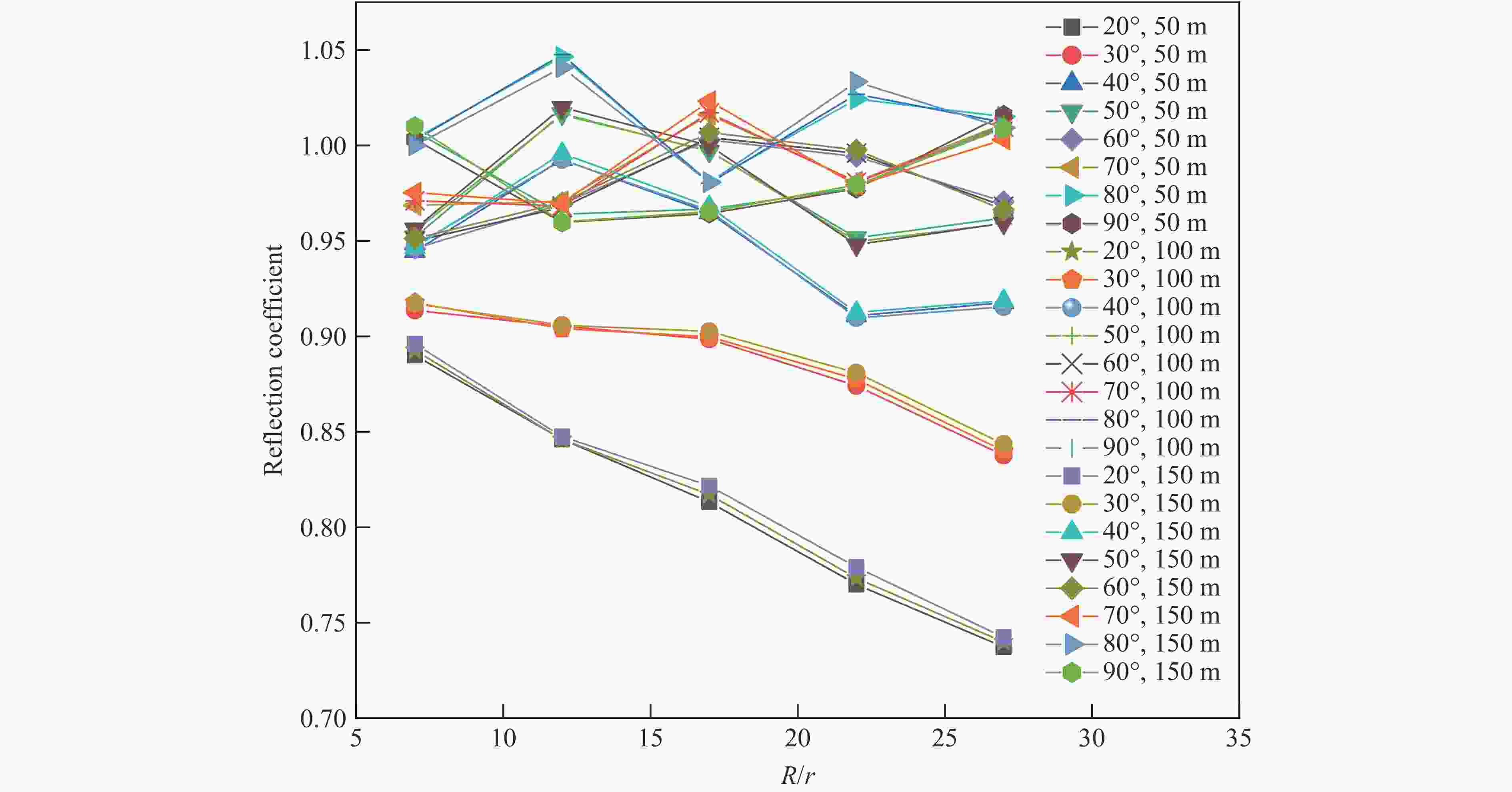
图 9 不同水深反射系数随测点角度及爆距比的变化关系
Figure 9. Relationship between reflection coefficient and explosion distance ratio in different water depths
ρ/(g·cm−3) D/(km·s−1) A/GPa B/GPa R1 R2 ω e/(kJ·g−1) 1.630 6.93 371.2 3.21 4.15 0.95 0.3 4.29  下载: 导出CSV
下载: 导出CSV
ρ/(g·cm−3) C0/(km·s−1) S Γ0 1.024 1.483 1.75 0.28
下载: 导出CSV
ρ/(kg·m−3) γ pa/MPa cV/(J·g−1·K−1) 1.17 1.4 0.10 1.012
下载: 导出CSV
Seafloor sediment ρ/(kg·m−3) E/MPa ν φ/(°) c/MPa Model 1 1.4 50 0.3 24 0.1
下载: 导出CSV
表 5 数值模拟与经验公式结果对比
Table 5. Comparison of numerical simulation and empirical formula results
R/r Peak pressure/MPa Error/% Simulation Empirical formula 7 205.03 193.72 5.84 12 96.32 86.90 10.84 17 60.65 58.62 3.45 22 43.04 43.81 1.75 27 32.58 37.91 14.07
下载: 导出CSV
表 6 数值模拟工况设置
Table 6. Settings of simulation cases
Case H/m Seafloor sediment Explosive environment 1 50 Nothing Free field 2 50 Model 1 Near the seabed 3 50 Model 2 Near the seabed 4 100 Model 1 Near the seabed 5 150 Model 1 Near the seabed
下载: 导出CSV
表 7 自由场与近海底测得的冲击波峰值压力对比
Table 7. Comparison of peak pressures of free-field and near-seabed underwater explosion shock wave
R/r α/(°) pmax/MPa Reflection
coefficientR/r α/(°) pmax/MPa Reflection
coefficientNear seabed Free field Near seabed Free field 7 20 179.29 201.44 0.89 12 60 92.73 95.67 0.97 7 30 186.21 203.80 0.91 12 70 95.00 97.86 0.97 7 40 193.43 204.80 0.94 12 80 98.33 93.98 1.05 7 50 194.98 204.80 0.95 12 90 92.42 96.30 0.96 7 60 192.78 203.80 0.95 17 20 47.78 58.76 0.81 7 70 195.10 201.43 0.97 17 30 53.24 59.25 0.90 7 80 202.46 201.74 1.00 17 40 56.52 58.58 0.96 7 90 196.41 195.79 1.00 17 50 58.40 58.58 1.00 12 20 82.82 97.87 0.85 17 60 59.44 59.27 1.00 12 30 86.62 95.67 0.91 17 70 61.07 60.10 1.02 12 40 93.96 94.66 0.99 17 80 60.00 61.17 0.98 12 50 96.23 94.65 1.02 17 90 57.77 59.91 0.96
下载: 导出CSV
-
[1] BLAKE J R, GIBSON D C. Cavitation bubbles near boundaries [J]. Annual Review of Fluid Mechanics, 1987, 19: 99–123. doi: 10.1146/annurev.fl.19.010187.000531 [2] 张永坤. 水下爆炸对沉底目标毁伤效应试验验证研究 [J]. 舰船电子工程, 2022, 42(5): 154–159. doi: 10.3969/j.issn.1672-9730.2022.05.034ZHANG Y K. Experimental research on the damage effects of bottom torpedoes shocked by underwater explosions [J]. Ship Electronic Engineering, 2022, 42(5): 154–159. doi: 10.3969/j.issn.1672-9730.2022.05.034 [3] 邵建军, 张永坤, 赵红光, 等. 基于相似理论的炸药海中沉底爆炸能量计算 [J]. 四川兵工学报, 2015, 36(6): 124–127. doi: 10.11809/scbgxb2015.06.031SHAO J J, ZHANG Y K, ZHAO H G, et al. Energy calculation of underwater explosion over seabed charge based on similarity law [J]. Journal of Sichuan Ordnance, 2015, 36(6): 124–127. doi: 10.11809/scbgxb2015.06.031 [4] 杨莉, 汪玉, 黄超, 等. 不同水底介质对有限域中装药沉底爆炸特性的影响 [J]. 高压物理学报, 2012, 26(5): 545–550. doi: 10.11858/gywlxb.2012.05.010YANG L, WANG Y, HUANG C, et al. Effects of different grounds on the loading characteristics of limited underwater explosion from a bottom charge [J]. Chinese Journal of High Pressure Physics, 2012, 26(5): 545–550. doi: 10.11858/gywlxb.2012.05.010 [5] 杨莉, 汪玉, 汪斌, 等. 沉底装药水中爆炸现象的实验研究 [J]. 爆炸与冲击, 2013, 33(2): 175–180. doi: 10.11883/1001-1455(2013)02-0175-06YANG L, WANG Y, WANG B, et al. Experimental investigation on loading characteristics of underwater explosion from a bottom charge [J]. Explosion and Shock Waves, 2013, 33(2): 175–180. doi: 10.11883/1001-1455(2013)02-0175-06 [6] 杨莉, 巨圆圆, 武堃, 等. 装药沉底爆炸峰值压力试验及数值模拟研究 [J]. 兵工学报, 2014, 35(Suppl 2): 368–372.YANG L, JU Y Y, WU K, et al. Experiment and simulation on peak pressure of underwater explosion of a bottom charge [J]. Acta Armamentarii, 2014, 35(Suppl 2): 368–372. [7] 杨莉, 汪玉, 杜志鹏, 等. 沉底装药水下爆炸冲击波传播规律 [J]. 兵工学报, 2013, 34(1): 100–104. doi: 10.3969/j.issn.1000-1093.2013.01.018YANG L, WANG Y, DU Z P, et al. Research on shock wave propagation of underwater explosion of bottom charge [J]. Acta Armamentarii, 2013, 34(1): 100–104. doi: 10.3969/j.issn.1000-1093.2013.01.018 [8] 黄潇, 张玮, 张阿漫. 近海底爆炸气泡对潜艇影响的研究 [J]. 中国造船, 2014, 55(3): 25–35. doi: 10.3969/j.issn.1000-4882.2014.03.003HUANG X, ZHANG W, ZHANG A M. Study on the effect of a grounding explosion bubble on submarine [J]. Shipbuilding of China, 2014, 55(3): 25–35. doi: 10.3969/j.issn.1000-4882.2014.03.003 [9] 姚熊亮, 杨文山, 陈娟, 等. 沉底水雷爆炸威力的数值计算 [J]. 爆炸与冲击, 2011, 31(2): 173–178. doi: 10.11883/1001-1455(2011)02-0173-06YAO X L, YANG W S, CHEN J, et al. Numerical calculation of explosion power of mines lying on seabed [J]. Explosion and Shock Waves, 2011, 31(2): 173–178. doi: 10.11883/1001-1455(2011)02-0173-06 [10] 邵宗战, 宋敬利, 贾则. 沉底水雷爆炸威力测量与评估方法研究 [J]. 水雷战与舰船防护, 2012, 20(1): 15–17, 46.SHAO Z Z, SONG J L, JIA Z. Research on the measurement and evaluation methods of the bottom mine’s explosive power [J]. Mine Warfare & Ship Self-Defence, 2012, 20(1): 15–17, 46. [11] 叶林征, 祝锡晶, 王建青, 等. 基于CEL不同角度超声空化微射流冲击的仿真分析 [J]. 振动与冲击, 2016, 35(16): 130–134, 157. doi: 10.13465/j.cnki.jvs.2016.16.021YE L Z, ZHU X J, WANG J Q, et al. Simulations of ultrasonic cavitation micro-jet impact with different angles based on CEL [J]. Journal of Vibration and Shock, 2016, 35(16): 130–134, 157. doi: 10.13465/j.cnki.jvs.2016.16.021 [12] 谭皓洋. 基于CEL方法的舰船近场水下爆炸全过程数值模拟研究 [D]. 哈尔滨: 哈尔滨工程大学, 2017.TAN H Y. Numerical simulation of the whole process of underwater explosion near warship based on CEL method [D]. Harbin: Harbin Engineering University, 2017. [13] AMBROSINI R D, LUCCIONI B M, DANESI R F, et al. Size of craters produced by explosive charges on or above the ground surface [J]. Shock Waves, 2002, 12(1): 69–78. doi: 10.1007/s00193-002-0136-3 [14] LUCCIONI B, AMBROSINI D, NURICK G, et al. Craters produced by underground explosions [J]. Computers & Structures, 2009, 87(21/22): 1366–1373. doi: 10.1016/j.compstruc.2009.06.002 -

 下载:
下载:









计量
- 文章访问数: 679
- HTML全文浏览量: 325
- PDF下载量: 52
