




-
摘要: 采用柱面冲击波回收装置,通过炸药爆轰产生的冲击波作用于Pb4O3、ZrO2和TiO2混合物粉体以合成PZT95/5粉体。通过对回收粉体进行的X射线衍射(XRD)分析,并结合冲击波理论,从实验和理论两个方面探讨了PZT粉体的合成机理和过程。结果表明,PZT的合成反应与Pb3O4的分解反应几乎同时进行,由于冲击波的特殊性,系统的温度和压力能同时满足Pb3O4分解和PZT合成的反应热力学条件,由Pb3O4分解的PbO一旦形成,就立刻与ZrO2、TiO2等氧化物反应生成PZT;冲击波合成PZT粉体属于特殊的固相反应,物质的扩散速度和反应速度大大提高。Abstract: A cylinder shock-wave recycling device was used, and via impacting effects of explosive shock waves, PZT95/5 powders were synthesized from a mixture of Pb3O4, ZrO2, and TiO2 oxides. Based on the XRD analyses of recovered powders and shock wave theory, the synthesis mechanism and process of PZT powders are discussed from the experimental and theoretical facets. The results show that PZT synthesis and Pb3O4 decomposition happened simultaneously, because of the shock-wave particularity, the system temperature and pressure could simultaneously satisfy the thermodynamics conditions of Pb3O4 decomposition and PZT synthesizing reactions. Thus once PbO formed by decomposing Pb3O4, PbO would react immediately with ZrO2 and TiO2 oxide compounds to produce PZT. And the reaction of shocksynthesizing PZT powders belonged to the special solid phase reaction, in which the diffusion velocity and reaction speed were enhanced greatly.
-
Key words:
- PZT95/5 powder /
- shock wave /
- mechanism of synthesis
-
轻质结构的抗冲击性能近年来得到了十分广泛和深入的研究,涉及的载荷有接触和非接触式的空气以及水下爆炸、高速破片侵彻和撞击等,对于其工程应用有重要的指导意义。碳纤维增强复合材料层合板凭借其高比强度、高比模量以及较好的隐身吸波性能,在航空航天领域以及快速响应舰船工业中已经取代了部分传统的金属材料和结构,成为现代三航工业领域不可或缺的一部分。
早期开展的纤维增强复合材料层合板结构动态失效行为研究工作主要以落锤撞击和弹体侵彻形成的接触式冲击加载为主[1-2]。研究结果表明,纤维增强复合材料层合板的主要失效模式包含基体和纤维断裂、层裂等。Hashin[3]指出,纤维和基体材料、铺层方式、几何尺寸和加载面积等均会对层合板的失效模式造成重要影响。Heimbs等[4]针对层合板的高速侵彻开展了实验和数值模拟,得出侵彻载荷下层合板除了纤维失效外,还包含层裂和基体开裂等。侵彻速度、侵彻角度、纤维铺层、层合板结构形式等对层合板结构在冲击载荷作用下的动态行为和失效均有较为丰富的研究成果[5-9]。Yang等[10]采用三维DIC对碳纤维编织层合板材料在弹体侵彻下的横向动态响应过程进行了研究。Li等[11]对编织的玄武岩/环氧树脂层合板的平板和曲面板结构在爆炸载荷作用下的动态失效行为进行了对比分析,强调了结构曲率对动态响应的重要影响。Huang[12]、Avachat等[13]研究了水下冲击载荷作用下层合板的动态行为和失效机理。Schiffer等[14]采用水下冲击加载模拟装置,对层合板在高强度水下冲击载荷作用下的响应进行研究,建立了复合材料层合板动态响应理论分析模型。为了降低对复杂空气爆炸实验的依赖,利用泡沫弹高速撞击形成的冲击载荷来模拟空气爆炸载荷是当前的一种常规有效手段[15-17]。目前这种加载方式下层合梁的动态响应和失效研究公开文献较少。
本工作针对碳纤维增强复合材料层合板在泡沫铝子弹撞击产生的冲击载荷加载下的动态响应和失效模式,利用高速摄影系统,研究层合板在不同冲击强度下的动态响应特性和抗冲击性能。
1. 实验方法
1.1 实验材料
本实验所用的层合板为T700碳纤维增强环氧树脂基复合材料,层合板所采用的铺层顺序为[0/90/0/90/0]2s,总厚度为2.4 mm。
T700碳纤维复合材料单层板的材料属性:纵向刚度E1 = 100 GPa,横向刚度E2 = 80 GPa,泊松比ν12 = 0.21,剪切模量G12 = 4 GPa,纵向拉伸强度XT = 2 100 MPa,纵向压缩强度XC = 700 MPa,横向拉伸强度YT = 42 MPa,横向抗压强度YC = 160 MPa,层间剪切强度S = 104 MPa,密度ρ = 1 500 kg/m3。
为了获得碳纤维增强环氧树脂基复合材料(CFRP/Epoxy)层合板在不同冲击强度下的动态响应特点与失效模式,本实验通过采用密度为460 kg/m3的泡沫铝子弹以不同速度进行加载。泡沫铝弹体的屈服强度σy = 3.4 MPa,平台应力σp = 2.16 MPa,压实应变εD = 0.74。泡沫铝弹体为
∅ 39.6 mm × 50 mm的圆柱体。层合板在实验前被切割成几何尺寸为240 mm × 42 mm的梁结构。1.2 实验装置
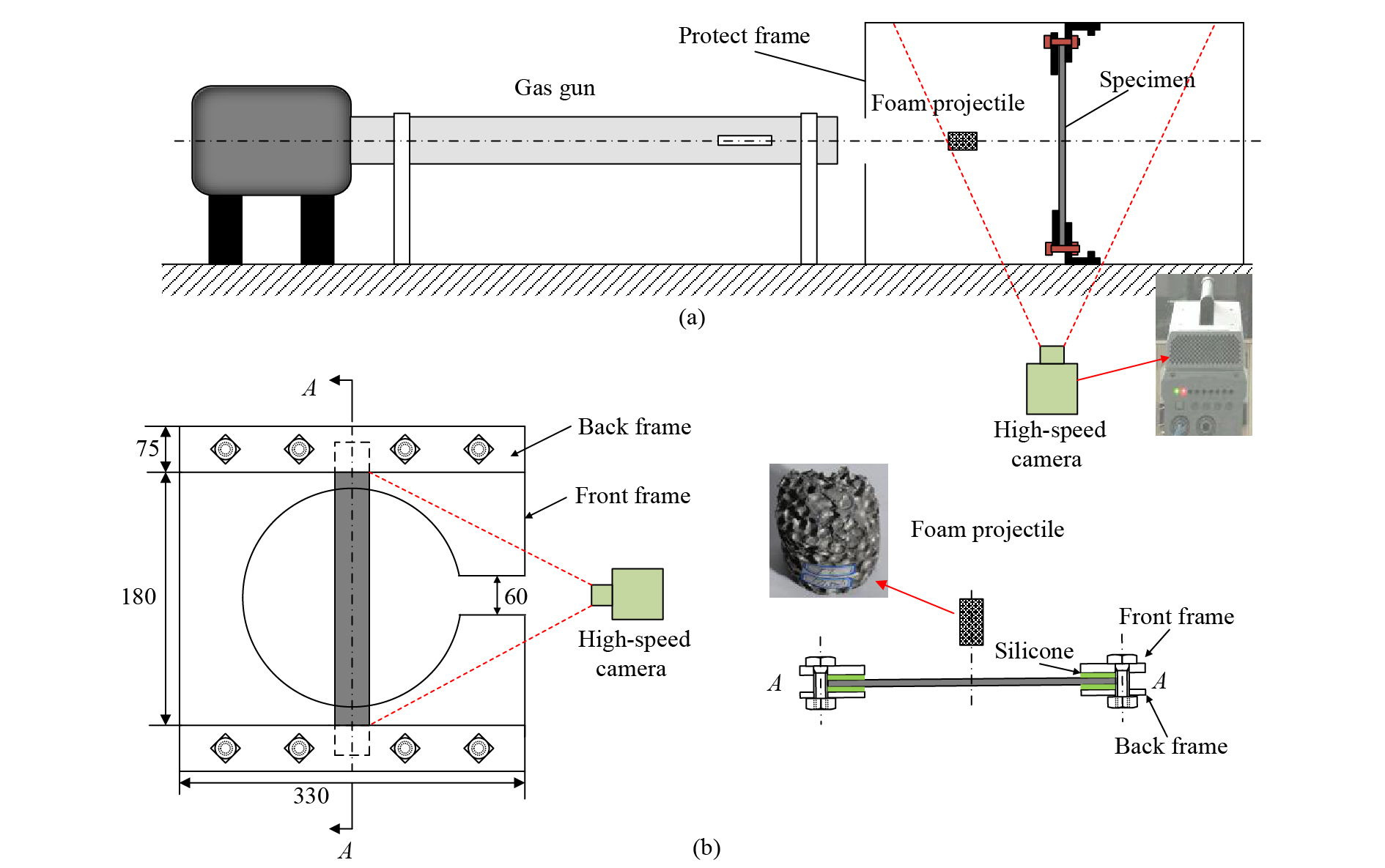
实验装置主要由一级轻气炮、激光测速装置、实验靶舱、固定支架以及高速相机等组成,如图1所示。通过控制一级轻气炮气室高压气体压力,达到控制泡沫弹初始速度的目的。实验过程中,利用Photron Fastcam Sa-Z高速摄影机捕捉泡沫弹冲击复合材料层合梁的全过程,高速相机采样频率为60 000帧每秒,分辨率为896 × 368像素。层合板梁采用螺栓固定的方式紧固于固定支架上。
 图 1 高速冲击加载实验装置(a)及固定夹具装置(b)示意图(单位:mm)Figure 1. Schematics of (a) the experimental set-up and (b) the clamped device (Unit: mm)
图 1 高速冲击加载实验装置(a)及固定夹具装置(b)示意图(单位:mm)Figure 1. Schematics of (a) the experimental set-up and (b) the clamped device (Unit: mm)1.3 实验工况
为了研究层合板梁动态响应与失效随冲击强度的变化,采用5种不同的弹体冲击速度,初始速度分别为50.1、71.4、138.8、173.5和204.2 m/s,对应的量纲一冲量(见下文)分别为0.24、0.33、0.67、0.83和0.99。
2. 实验结果与讨论
与金属梁结构在局部冲击载荷下形成的两个动态塑性铰链不同,复合材料层合板受冲击后无塑性变形发生。层合板的横向变形是在弯曲波的驱动下发生的,这种随着弯曲波传播而发生的横向变形过程在一定冲击强度下与塑性铰的运动形式基本相似[12]。然而,无塑性变形的碳纤维层合板的冲击响应也必然与金属梁的响应存在不同。
泡沫弹加载形成的初始冲量为
I0=mv0 ,其中m为泡沫弹质量,v0为弹体初始速度。本研究采用量纲一冲量¯I=103I=103I0/L√ρσ ,其中ρ和σ分别为层合板的密度与拉伸强度,L为梁的半跨长度。碳纤维增强层合板由于无剩余变形,结构在冲击变形过程中的横向变形是评估其抗冲击性能的重要参数。2.1 层合板失效模式和机理
在金属铝泡沫弹的局部冲击载荷加载下,CFRP/Epoxy层合板随着冲击强度的变化会发生一系列的变形和失效。由于边界简支,层合板梁的中心加载区域受到由弯曲/拉伸导致的最大应力。柱形泡沫弹在与层合板梁接触的边缘由于入射压缩应力波和弯曲波的作用会形成局部褶皱和横向变形。压缩应力波经由层合板背面反射形成拉伸波,当拉伸波强度足够大时,层合板出现纤维与基体之间的层裂。在足够大的初始冲击强度下,随着横向变形和轴向拉伸的增加,层合板发生基体和纤维的断裂失效。
2.2 层合板梁动态响应过程
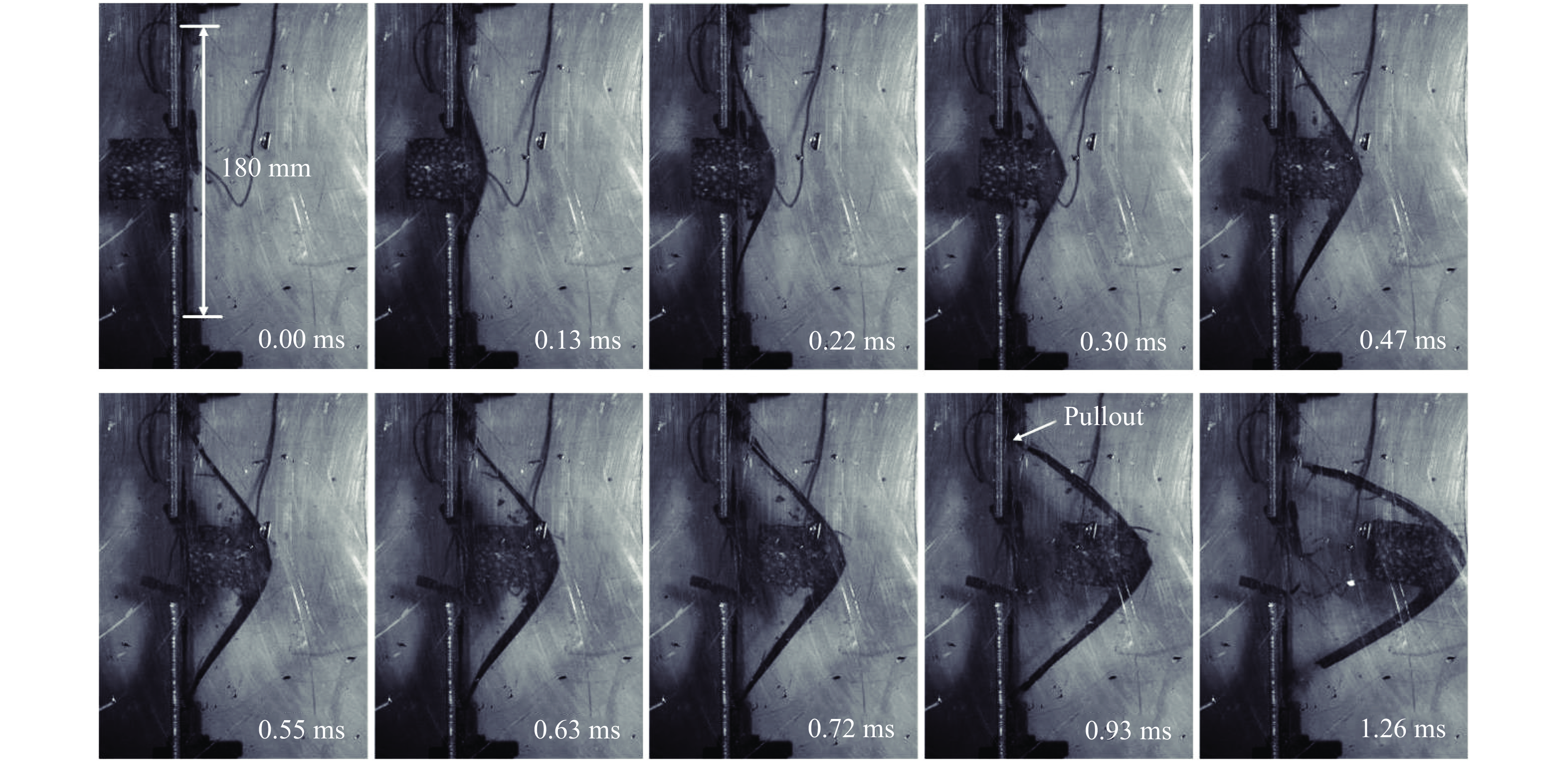
以初始冲击速度为138.8 m/s、量纲一冲量为0.67的泡沫铝弹体的冲击加载为例,图2展示了泡沫弹加载过程中层合板梁结构的动态变形和失效过程。整个变形过程可以分为两个典型阶段:(1)由中心局部加载形成的弯曲波向两端传播,直至到达端点处;(2)层合板在拉伸/弯曲下开始发生结构整体变形。这与金属梁受局部载荷作用的典型响应阶段相同。包括后续脱离靶架的运动过程,整个响应阶段的持续时间大约为2.00 ms。由图2可见,弯曲波在0.30 ms达到两端后,层合板的变形轮廓在弹体两侧呈直线,直到0.80 ms后脱离靶架。这种直线的变形在固定端约束的情况下,使得层合板在中心弹体加载位置发生明显的对折现象,进而导致基体和纤维拉伸断裂的发生。在1.26 ms,层合板已发生明显的对中折断。
 图 2 CFRP/Epoxy层合板梁在
图 2 CFRP/Epoxy层合板梁在¯I = 0.67冲击下的动态响应过程Figure 2. Sequence of high-speed photographs of the CFRP/Epoxy laminate subjected to¯I = 0.67层合板在变形过程中的变形呈较好的对称性,以下半跨长度为L的层合板上不同位置点随时间变化绘制层合板变形轮廓时程曲线,如图3(a)所示。可以清晰地看到冲击载荷作用下随着弯曲波的运动,层合板从局部变形到结构整体变形的过程。结合图2可以发现,由于层合板梁变形轮廓呈近似直线,在弹体装置位置未与弹体头部完全贴合时,整个变形在弯曲波到达左侧端点后开始以中点和端点两点为“铰”,发生轴向拉伸,最终发生对折失效。图3(b)所示的中点随时间的变形过程中可见在0.47 ms前是线性增长,在0.47 ms后由于固支端的作用,曲线斜率有所下降,直至0.68 ms开始逐步脱离夹具,随弹体向靶舱壁运动。
 图 3 CFRP/Epoxy层合板梁在
图 3 CFRP/Epoxy层合板梁在¯I = 0.67冲击下的变形轮廓(a)和中点变形(b)Figure 3. Histories of deformation profiles (a), and midpoint deflection (b) of the CFRP/Epoxy laminate subjected to¯I = 0.67图4为上述泡沫弹撞击下碳纤维/环氧树脂层合板梁的失效模式。层合板从中间断裂为两等分的同时,可以看到在中心撞击位置处层合板发生了明显的沿厚度方向的压缩失效。与此同时,泡沫弹撞击端也有一定的压缩发生。而泡沫弹端部的严重不对称压缩的主要原因是加载过程中弹体的轻微偏转以及以此姿态发生的与靶舱缓冲垫的二次撞击,如图2所示。从图4(c)所示的齐整断裂面可以看到明显的基体和纤维脆性断裂、层裂、基体裂纹和纤维拨出等失效模式,并且在加载区域两侧,由于层合板的弯曲,也出现了明显的层裂失效。
 图 4 CFRP/Epoxy层合板梁在
图 4 CFRP/Epoxy层合板梁在¯I = 0.67冲击下的失效模式Figure 4. Failure mode of the CFRP/Epoxy laminate subjected to¯I = 0.672.3 加载强度的影响
为了研究不同加载强度对层合板动态响应过程和抗冲击性能的影响,本研究开展了另外4组不同初始冲击强度加载的实验研究。泡沫弹体质量基本相同,冲击初始速度和量纲一加载强度如上所述。图5为不同初始冲击强度下层合板梁的动态变形和失效过程,呈现出明显不同的变形机制:低于中等冲击强度(v0 = 138.8 m/s)加载时,层合板梁的变形以结构整体的横向变形为主;高于中等冲击强度时,层合板的变形出现明显的局部化,并且局部化区域随着加载强度的增加而减小。出现这种局部化效应的主要原因可以归结为在强冲击载荷作用下碳纤维层合板的横向变形速度大于弯曲波在层合板中沿径向的传播速度。在较低冲击强度加载下,层合板随着整体变形的进行,在泡沫弹作用下呈现明显的平台,如图5(a)中0.43 ms图像,并随着变形的持续增加,最终呈整体圆拱状。强冲击作用下,层合板在发生局部明显横向变形的初始时刻便发生了明显的基体和纤维断裂等失效,尤其当弹体冲击速度为204.2 m/s时,在0.33 ms时便出现清晰的纤维基体破碎,在0.73 ms时基本完全失效。
 图 5 不同冲击强度下层合板动态变形和失效Figure 5. Dynamic deformation and failure of CFRP/Epoxy laminate under different impulses
图 5 不同冲击强度下层合板动态变形和失效Figure 5. Dynamic deformation and failure of CFRP/Epoxy laminate under different impulses图6(a)对比了最低和最高两种冲击强度下的变形轮廓线随时间变化关系,显示了明显的变形局部化的特点。当弹体以204.2 m/s速度冲击时,层合板两侧部分始终沿着径向滑移,未发生横向变形;在距离中点20~50 mm段的变形较小;在靠近中心的40 mm段则发生急剧的变形增长。低速冲击时,在0.13 ms之后,结构发生整体变形。取不同冲击强度下层合板的中点变形,得到如图6(b)所示的时程曲线。随着结构变形的增加,最终靶板脱离夹具,继续以一定的动能运动(见图5,以空心点的形式表示)。随着冲击强度的增加,曲线斜率明显增加,中点响应速度增加而靶板脱离夹具的时间减小。这种冲击强度的影响在冲击速度为50.1~138.8 m/s时明显地大于冲击速度为138.8~204.2 m/s。当冲击强度较小时,结构整体响应时间长,并在停止前发生了较明显的变形恢复阶段。当冲击强度增加时,随着响应速度的增加和局部化失效的发生,层合板的最大变形在138.8 m/s时达到最大的67.2 mm后开始降低。
 图 6 层合板在不同冲击强度下的中点变形(a)和变形轮廓线(b)Figure 6. Midpoints-deflection histories (a) and deformation profiles (b) of CFRP laminates under impulsive loadings
图 6 层合板在不同冲击强度下的中点变形(a)和变形轮廓线(b)Figure 6. Midpoints-deflection histories (a) and deformation profiles (b) of CFRP laminates under impulsive loadings当弹体冲击速度小于138.8 m/s时,层合板主要发生弹性变形,在层合板表面无明显失效发生。在弹体冲击速度达到或超过138.8 m/s时,层合板随冲击强度的增加,发生折断失效。速度到达204.2 m/s时层合板完全破碎,如图7所示。
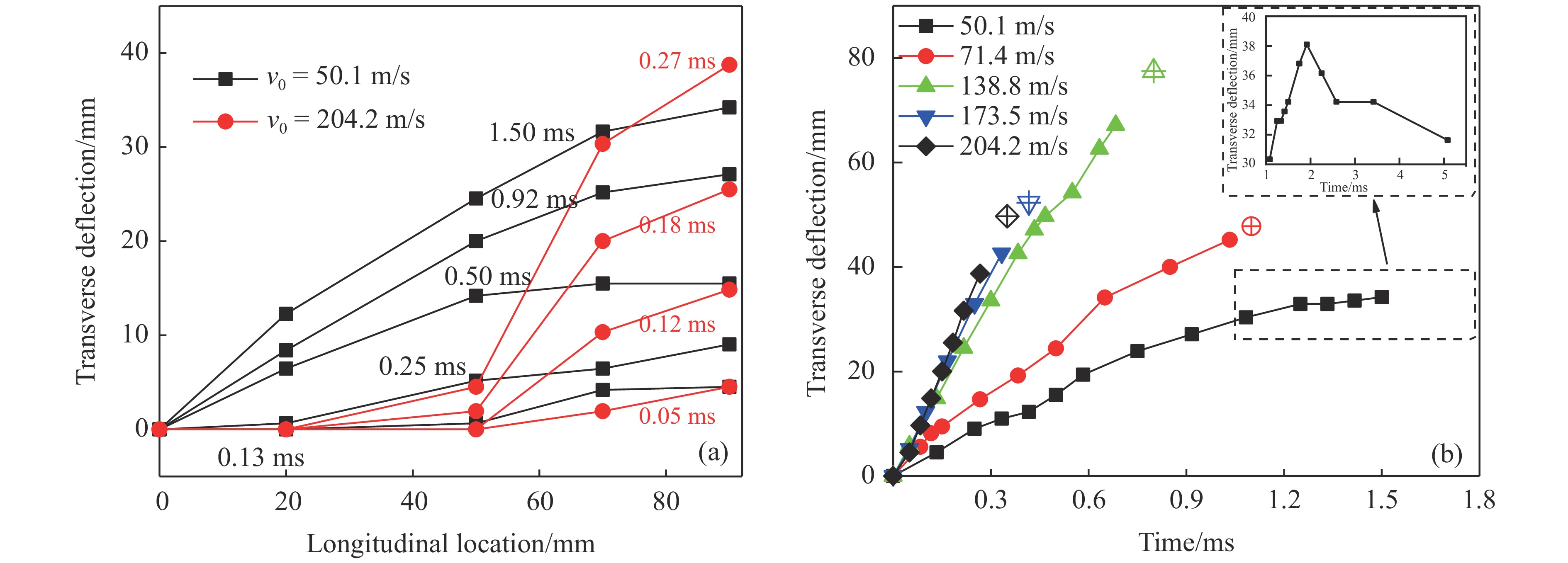
 图 7 CFRP/Epoxy层合板失效模式与能量耗散比的关系Figure 7. Failure modes versus specific energy absorption of the CFRP/Epoxy laminate
图 7 CFRP/Epoxy层合板失效模式与能量耗散比的关系Figure 7. Failure modes versus specific energy absorption of the CFRP/Epoxy laminate2.4 层合板能量吸收
由获得的动态变形过程中的图像可以发现,泡沫弹在与层合板撞击过程中的变形较小,其主要变形发生在与靶舱的二次碰撞。因此,本研究忽略泡沫弹的变形吸能。层合板在泡沫弹冲击加载下的能量耗散方式主要有结构变形和层合板失效两种。层合板的耗能Ed可以通过弹体初始动能E0和靶板与弹体脱离夹具时获得的剩余动能Er之差来获取。在实验过程中,靶板在拔出之前与固支部分及夹具发生摩擦,但是由于作用时间短,做功相较于前后动能很小,本研究将此忽略。在弹体冲击下,靶板和弹体同步向初始速度方向运动,在不考虑靶板速度沿跨长分布的情况下,假定在脱离靶板后,层合板和泡沫弹以相同速度向前运动。因此,
Ed=E0−Er 。其中剩余能量Er=(1/2)mtv2r ,mt 和vr 是靶板和弹体的总质量和共同速度。而碳纤维层合板材料的能量耗散比可记为SEA=Ed/ρ 。如图7所示,层合板能量耗散比随初始冲击强度增加呈上升趋势,并呈现出明显的3个阶段,表现出与层合板失效模式的直接对应关系。在低速撞击情况下,层合板发生以弹性变形为主的失效,能量耗散比上升较快;随着中心折断失效的发生,能量耗散伴随着更大面积的基体和纤维断裂而有所增加,但增加幅度明显降低;在最大冲击强度下,层合板整体在初始撞击时刻便发生严重的大面积失效,致使能量耗散有了明显的增加。3. 结 论
采用泡沫铝子弹撞击形成的冲击载荷,对等厚度的碳纤维增强复合材料层合板的抗冲击性能展开了实验研究,讨论了冲击强度对层合板的动态失效过程、变形轮廓、中点变形、失效模式及能量耗散比的影响。
(1)低于该临界加载强度时,层合梁的变形以整体的横向变形为主,在弹体撞击位置呈较为明显的平台;高于该临界加载强度时,层合梁的变形发生明显的局部化,并且局部化区域随着加载强度的增加而减小。
(2)中点的变形响应速度随冲击强度的增加而增加。随着响应速度的增加和局部化失效的发生,层合梁临界冲击速度下的最大变形开始降低。层合梁的弹性最大变形不能作为一个独立的抗冲击性能评价参数。
(3)随着冲击强度的增加,层合梁的失效主要分为低速冲击下的弹性变形、中等强度冲击下的对折断裂以及高强度冲击下的完全破碎。层合梁能量耗散比随冲击强度的增加而增加,并展现出与这3类结构失效模式直接关联的3个不同阶段。
-
Thadhani N N. Shock-Induced Chemical Reactions and Synthesis of Materials [J]. Progress in Materials Science, 1993, 37: 117-226. Kuznetsova E M, Reznichenko L A, Razumovskaya O N, et al. Shockwave Activation of High-Temperature Ferroelectric Powders [J]. Technical Physics Letters, 2000, 26(9): 767-770. Liao Q L, Yang Sh Y, Cai L C, et al. Synthesis of Hydroxyapatite Powder by Shock Wave Treatment Method [J]. Chinese Journal of High Pressure Physics, 2002, 16(4): 249-253. (in Chinese) 廖其龙, 杨世源, 蔡灵仓, 等. 用冲击波合成法制备羟基磷灰石粉体 [J]. 高压物理学报, 2002, 16(4): 249-253. [4] Liu J J, Tan H, Xu K, et al. Shock Synthesis of Zinc Ferrite and Its Photocatalytic Activity in Dehydrogenation of H2S [J]. Chinese Journal of High Pressure Physics, 1997, 11(2): 90-97. (in Chinese) 刘建军, 谭华, 徐康, 等. 纳米铁酸锌的冲击波合成及它的光催化活性 [J]. 高压物理学报, 1997, 11(2): 90-97. Jia L G, Xiong D Y. Influencing Factors of TiC Synthesis by Shock Wave [J]. Nonferrous Metal, 2002, 54(4): 1-5. (in Chinese) 贾丽改, 熊代余. 冲击波方法合成TiC的影响因素 [J]. 有色金属, 2002, 54(4): 1-5. Wang J X, Yang Sh Y, He H L, et al. Structure and Properties of Lead Zirconate Titanate 95/5 Powders Synthesized by Shock Wave Technique [J]. Journal of the Chinese Ceramic Society, 2005, 33(6): 718-722. (in Chinese) 王军霞, 杨世源, 贺红亮, 等. 冲击波合成Pb(ZrTi0. 05)O3粉体的结构和特性 [J]. 硅酸盐学报, 2005, 33(6): 718-722. do Ian P H , Benson D J. Micromechanical Modeling of Shock-Induced Chemical Reactions in Heterogeneous Multi-Material Powder Mixtures [J]. International Journal of Plasticity, 2001, 17: 641-668. Gong P, Tang Zh P, Shen Zh W. Experimental Investigation and DEM Simulation of Mass Mixing under Shock Loading [J]. Chinese Journal of High Pressure Physics, 2004, 18(1): 21-26. (in Chinese) 龚平, 唐志平, 沈兆武. 冲击下材料质量混合的实验研究及离散元模拟 [J]. 高压物理学报, 2004, 18(1): 21-26. Zemsky S V, Ryabchikov Y A, Epshteyn G N, et al. Mass Transfer of Carbon under the Influence of a Shock Wave [J]. Phys Met Metall, 1979, 46: 171-173. Yang Ch, Hu J B. Deffusion of Tungsten Atom in Iron and Nickel under the Shock Loading [J]. Ordnance Material Science and Engineering, 1997, 20(2): 20-23. (in Chinese) 杨超, 胡金彪. 冲击载荷下钨在铁和镍中的扩散 [J]. 兵器材料科学与工程, 1997, 20(2): 20-23. Zhang W J. A Discussion on the Mechanism of Shock-Induced Transformation of Graphite to Diamond [J]. Chinese Journal of High Pressure Physics, 2004, 18(3): 217-227. (in Chinese) 张万甲. 冲击引起石墨金刚石相转变机理的探讨 [J]. 高压物理学报, 2004, 18(3): 217-227. Li X J, Wang J X, Zhang Y J, et al. Research of Temperature Rise at the Particles Interface Caused by Adiabatic Friction in Explosive Consolidation of Powders [J]. Chinese Journal of High Pressure Physics, 2004, 18(2): 97-102. (in Chinese) 李晓杰, 王金相, 张越举, 等. 爆炸粉末烧结颗粒间摩擦引起的界面温升研究 [J]. 高压物理学报, 2004, 18(2): 97-102. Li Sh P. Processes for Advanced Ceramics [M]. Wuhan: Wuhan University of Technology Press, 1990: 202-203. (in Chinese) 李世普. 特种陶瓷工艺学 [M]. 武汉: 武汉工业大学出版社, 1990: 202-203. -


 下载:
下载:
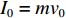
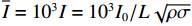

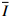

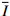
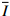

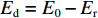
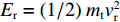
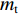



 点击查看大图
点击查看大图
计量
- 文章访问数: 9736
- HTML全文浏览量: 608
- PDF下载量: 876
 下载:
下载:
